[box type=”bio”] What to Learn from this Article?[/box]
Despite obvious clinical findings, thorough radiographic evaluation should be done to determine full extent of the disease & future complications. Treatment requires multidisciplinary approach.
Case Report | Volume 5 | Issue 2 | JOCR April-June 2015 | Page 21-24| Purva Prakash Patil, Suresh Ramchandra Barpande, Jyoti Dilip Bhavthankar, Jayanti G. Humbe. DOI: 10.13107/jocr.2250-0685.264
Authors: Purva Prakash Patil[1], Suresh Ramchandra Barpande[1], Jyoti Dilip Bhavthankar[1], Jayanti G. Humbe[1]
[1] Department of Oral Pathology & Microbiology, Government Dental College & Hospital, Dhanwantari Nagar, Ghati Campus, Aurangabad, Maharashtra, India-431001.
Address of Correspondence:
Dr. Purva Prakash Patil,
B.D.S., Postgraduate Student,133, Department of Oral Pathology, Government Dental College & Hospital, Dhanwantari Nagar, Ghati Campus, Aurangabad, Maharashtra, India-431001.
E-mail: patilpurvap@gmail.com
Abstract
Introduction: Cleidocranial dysplasia (CCD) is characterized by aplasia or hypoplasia of the clavicles, characteristic craniofacial malformations, and the presence of numerous supernumerary and unerupted teeth. It affects bones derived from both intra-membranous and endochondral ossification. Incidence has been reported as 1 in 10,00,000. It is caused by mutation in the gene encoding transcription factor Core Binding Factor Subunit Alpha 1 (CBFA1) or Runt related transcription factor 2 (RUNX2).
Case Report: This presentation discusses the clinical and radiographic features of a familial case of cleidocranial dysplasia occurring in a father and a child. All the clinical and radiographic features, except that of the chest x-ray, were more prominent in the child than the father.This supports the fact that CCD is transmitted by an autosomal-dominant mode of inheritance with high penetrance and variable expressivity. It is sporadic in about 40% of cases. Each child of an individual with CCD has a 50% chance of inheriting the mutation.
Conclusion: Diagnosis is mostly made on the basis of clinical and radiographic features. Molecular genetic testing such as sequence analysis or deletion analysis can be used in cleidocranial dysplasia. Some cases are diagnosed through incidental findings by physicians, treating patients for unrelated conditions. Treatment of these patients requires a multidisciplinary approach which includes orthopaedic and dental corrections along with management of any complications of cleidocranial dysplasia.
Keywords: Cleidocranial dysplasia, Coxa vara, Core Binding Factor Subunit Alpha 1 (CBFA1), wormian bones, pseudoepiphyses.
Introduction
Cleidocranial dysplasia (CCD) is skeletal dysplasia characterized by aplasia or hypoplasia of the clavicles, characteristic craniofacial malformations, and the presence of numerous supernumerary and unerupted teeth [1]. It is also known as Marie and Sainton’s disease, mutational dysostosis or cleidocranial dysostosis [2]. The first case of clavicular defects was reported by Martin in 1765 [3]. Cleidocranial dysplasia was first described by Pierre Marie and Paul Sainton in 1898. Since then, over 1000 cases have been documented in the medical literature [1, 2]. Hesse was the first to describe in detail the defects of dentition and jaws associated with cleidocranial dysplasia [3]. This article reports a case of CCD with a familial occurrence in a father and child reported to our institute & also discusses the complete spectrum of clinical and radiographic features in CCD.
Case report
Report 1
A 19-year-old male patient reported with a main complaint of pain in the upper right first molar. He was (Fig. 1) short in stature and had drooping shoulders. The facial appearance was frontal bossing, mild hypertelorism, mid-face deficiency and relative mandibular prognathism. His intelligence was normal. Family history revealed similar findings in his father, presented here as case 2. Siblings were unaffected. Intra-oral examination (Fig.2) revealed a high arched palate, unilaterally constricted maxillary arch with malaligned multiple over-retained deciduous teeth and few erupted permanent teeth. The clinical features were suggestive of CCD. 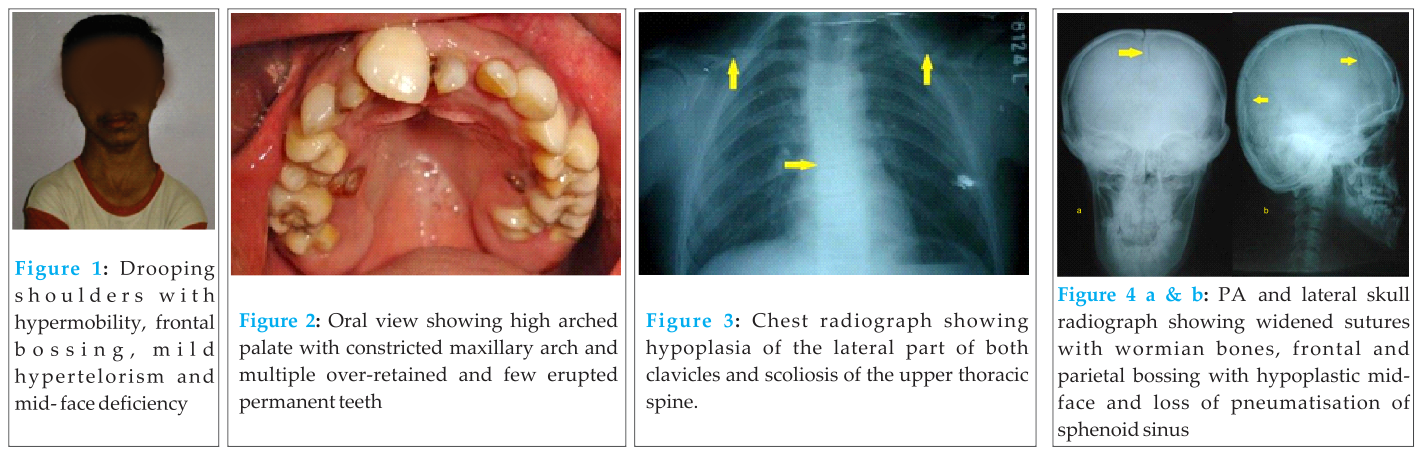
The skull, jaw, chest, pelvis and hand and wrist radiographs were evaluated. The chest x-ray (Fig.3) showed hypoplasia of the lateral part of both clavicles and scoliosis of the upper thoracic spine. Widened cranial sutures with wormian bones, loss of pneumatisation of sphenoid sinus, frontal and parietal bossing, and hypoplastic mid-facial region were evident on skull radiographs (Fig.4). The panoramic view (Fig.5) of the jaws showed multiple impacted teeth, over-retained deciduous teeth, hypoplastic maxillary sinus and slender coronoid processes. An abnormal pelvis shape and coxa vara were seen on the pelvis radiograph (Fig.6) whereas the hand and wrist radiographs (Fig.7) showed slender, elongated metacarpals and phalanges and tapering distal phalanges. The diagnosis of CCD was confirmed.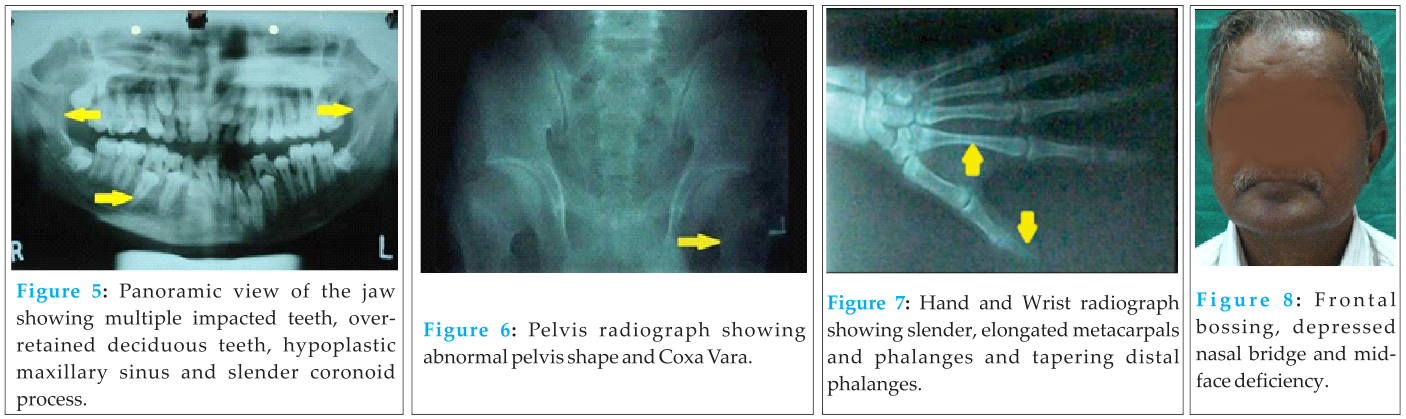
Report 2
Case 1 patient was accompanied by his father who was 50 years old with (Fig.8) short stature and drooping shoulders. He was also able to approximate his shoulders anteriorly. Family history revealed unaffected parents and siblings. His facial appearance was frontal bossing, depressed nasal bridge, mid-face deficiency, reduced lower facial height and relative mandibular prognathism. Intra-orally, few teeth were present (Fig.9). There was a history of extraction of all erupted teeth 25 years previously followed by complete denture prosthesis. The present teeth had erupted 4 years previously. A moderately narrow palate was also seen. 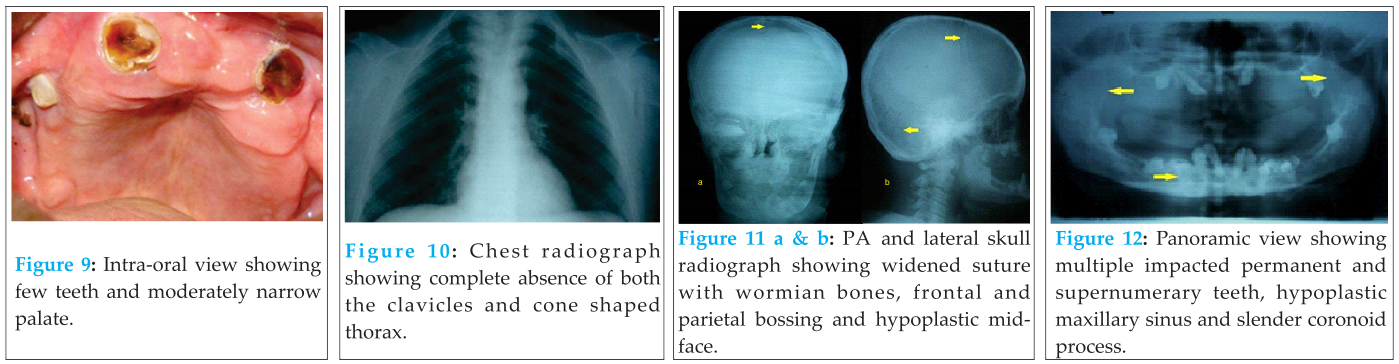
Detailed radiographic evaluation exhibited skeletal deformities similar to those of his son except that he had complete absence of clavicles bilaterally and a more cone shaped thorax on the chest x-ray (Fig.10). Skull radiographs (Fig.11) revealed widened sutures with wormian bones, frontal and parietal bossing. There was hypoplasia of the sphenoid bones and mid-facial region. The panoramic radiograph (Fig.12) showed multiple impacted permanent and supernumerary teeth. Hypoplastic maxillary sinus and slender coronoid processes were also evident. Pelvic changes (Fig. 13) were mild with minimal narrowing of the pelvic inlet and mild grade coxa vara. Hand and wrist radiographs (Fig. 14) showed thinning of the cortex and widening of medullary cavity of the proximal phalanges and slender elongated second metacarpals.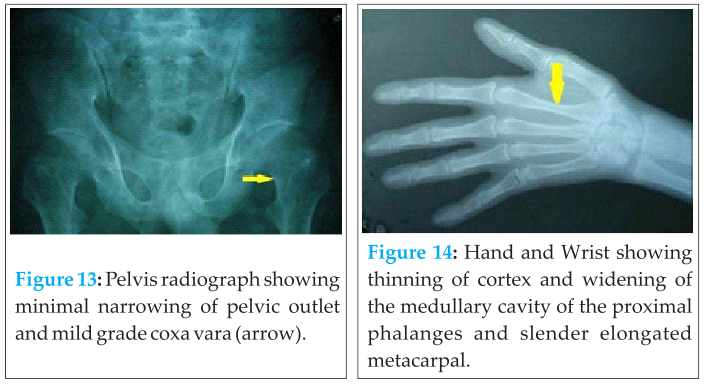
The above clinical and radiological features in both cases confirmed the diagnoses of cleidocranial dysplasia.
Discussion
Cleidocranial dysplasia is rare in occurrence with an incidence of 1:10,00,000 [2]. It is caused by mutation in the gene on 6p21 encoding transcription factor Core Binding Factor Subunit Alpha 1 (CBFA1) or Runt related transcription factor 2 (RUNX2) [1]. RUNX2 is a part of the Fibroblast Growth Factor (FGF) and Bone Morphogenetic Protein (BMP) signaling pathways in tooth and bone development respectively [4]. FGF and BMP4 induce both Msx1-dependent and Msx1-independent signalling pathways in early tooth development. RUNX2 is a major regulator of differentiation and maturation of osteoblasts and chondrocytes [5]. Here, both father and son presented with similar skeletal and dental abnormalities. All clinical and radiographic features except that of the chest x-ray, were more prominent in the child than the father. This supports the fact that CCD is transmitted by an autosomal-dominant mode of inheritance with high penetrance and variable expressivity.1 Sporadic occurrence has been reported in about 40% of cases [6] and autosomal recessive inheritance has also been reported [7]. In the cases reported here, it appears that CCD occurred sporadically in the father, which was then transmitted by autosomal dominant mode of inheritance to the child. Diagnosis is mostly made on the basis of clinical and radiographic features. Molecular genetic testing such as sequence analysis or deletion analysis can be applied in CCD for genotypic evaluation. Mutations have been detected in 60%-70% of individuals with clinical diagnosis of CCD [8].
The phenotypic spectrum of CCD ranges from mildly affected individuals with dental anomalies only, to severely affected patients with syringomyelia [6]. Though bones formed by endochondral ossification can also be affected, CCD most predominantly affects bones derived from intramembranous ossification, such as the cranium and the clavicles [8]. Individuals are usually short in stature. This is a proportionated dwarfism. The most striking skeletal defects are hypoplastic or aplastic clavicles, late closure of the fontanelles, open skull sutures and multiple wormian bones [2]. Partial hypoplasia commonly involves the acromial end of the clavicles [7]. Complete absence of clavicles is seen in 10% of cases as seen here in Case 2 [1]. The thoracic cage is small and bell or cone shaped with short, oblique ribs making the individual prone to chest infections. Scapulae are sometimes hypoplastic [7]. Muscle attachments to the clavicles may also be dysplastic, leading to distortion of the neck [3].
Cranial abnormalities include wide-open sutures, patent fontanelles, and the presence of wormian bones as seen in both reported cases. Delayed closure of cranial sutures and fontanelles leads to frontal, parietal and occipital bossing. Additionally, there may be poor or absent pneumatisation of paranasal, frontal and mastoid, sphenoid sinuses [9]. Facial features include depressed nasal bridge, hypertelorism with possible exophthalmos, hypoplastic mid-facial region and relatively prognathic mandible. The palate is often high arched [9].
Pelvic features are delayed ossification with wide pubic symphysis, hypoplastic iliac wings, widened sacroiliac joints and a large femoral neck resulting in coxa vara (a condition in which the angle between the head and neck of the femur and its shaft is decreased, usually defined as ≤ 120º). Hand and wrist radiographs show pseudoepiphyses of the metacarpal and metatarsal bones resulting in characteristic lengthening of the second metacarpal. Hypoplastic and pointed digital phalanges can also be seen [9].
Dental changes occur frequently and are very characteristic of CCD. Retention of the deciduous dentition with delayed eruption of the permanent teeth is a relatively constant finding. Delayed eruption of permanent teeth is probably attributed to generalized reduction in bone resorption and paucity of cementum [10]. The presence of multiple supernumerary teeth has been hypothesized to result from incomplete or delayed resorption of the dental lamina [3]. Supernumerary teeth were absent in Case 1 whereas Case 2 showed three supernumerary teeth in lower jaw. Morphologically, supernumerary teeth resemble their normal counterparts [7]. Hypoplasia of the masseter muscles is another feature, which may be due to discontinuity of the zygomatic arch. Hypofunction of the masseter muscle eventually leads to compensatory hyperfunction of the temporal muscles. This makes the anterior border of the mandibular ramus parallel to the posterior border, and the coronoid process is directed upwards and backwards in such cases [11].
The differential diagnosis of CCD includes Crane-Heise syndrome, mandibuloacral dysplasia, pycnodysostosis, Yunis-Varon syndrome, CDAGS syndrome (Craniosynostosis, anal anomalies, and porokeratosis) and hypophosphatasia etc. These conditions may share some characteristics with CCD; however, all these are autosomal recessive disorders and have other specific features [8].
Crane-Heise syndrome is characterized by a large head, poorly mineralized calvarium, cleft lip and palate, low-set dysplastic ears, hypoplastic clavicles and scapulae, agenesis of some cervical vertebrae, and genital hypoplasia [8].
Mandibuloacral dysplasia is a progressive disorder characterized by short stature, delayed closure of cranial sutures and dysplastic clavicles with mandibular hypoplasia. The scalp hair becomes sparse by the third decade and some individuals develop alopecia. The joints become progressively stiff; radiographs reveal acro-osteodysplasia of the fingers and toes, with delayed ossification of the carpal bones. Osteolysis of the mandibular body and ramus results in micrognathia. In adolescence, dental crowding is observed. Hypoplastic roots lead to early tooth loss. The skin is atrophic with decreased subcutaneous fat. Several individuals develop a hyperpigmented rash and hyperkeratotic papular lesions over the trunk and extremities respectively [8].
Pycnodysostosis is characterized by short stature, osteopetrosis, short terminal phalanges, and failure of closure of the cranial sutures and fontanelles. Radio-opacity of all bones is increased because of increased density of the trabecular bone but not the cortices [8].
Yunis Varon syndrome is characterized by prenatal growth deficiency, wide-open fontanelles and sutures, unusual mineralization of the skull, and hypoplastic clavicles. The thumbs and great toes are hypoplastic or absent [8].
CDAGS syndrome is characterized by craniosynostosis, delayed closure of the fontanelles, cranial defects, clavicular hypoplasia, anal and genitourinary malformations, and skin eruption [8].
Hypophosphatasia is characterized by a generalized defect of mineralization with delayed ossification of multiple skeletal elements. Children with the infantile form may present with very poorly mineralized cranium, widened cranial sutures, short ribs, and a narrow thorax. The alkaline phosphatase activity in serum and tissues is very low [8].
Common complications of CCD reported are pes planus, genu valgum, shoulder and hip dislocation, recurrent sinus infections, upper airway complications, recurrent ear infection, hearing loss, dental caries, osteomyelitis of the jaw bones and respiratory distress in early infancy which may be experienced because of narrow upper thoracic diameter [11].
Treatment is often directed at orthopaedic correction which includes correcting multiple dislocated shoulders, radial head or hips. If bone density is below normal, treatment with calcium and vitamin D supplementation should be considered [1]. To correct dental complications, Pusey and Durie suggested removal of only the erupted teeth and use of removable prosthesis to minimize alveolar bone loss [12]. However, subsequent eruption of retained teeth may require further surgery and modification of the prosthesis [1]. Each child of an individual with CCD has a chance of inheriting the mutation. Hence, it would be appropriate to offer genetic counselling to young adults who are affected [8].
Conclusion
The clinical findings of CCD are present at birth. But, they are often diagnosed at a much later time. Most cases are diagnosed as an incidental finding by physicians and dentists while treating patients for unrelated conditions. Family history, pathognomonic clinical and radiographic findings play a central role in the diagnosis of CCD. Treatment of these patients requires a multidisciplinary approach which includes orthopaedic and dental corrections along with management of any complications of CCD.
Clinical Message
Despite the obvious clinical findings, thorough radiographic evaluation is important to predict the full extent of the disease and potential complications. Treatment should not only include orthopaedic and dental corrections, but requires a multidisciplinary approach.
References
1. Toptanci IR, Colak H, Koseoglu S. Cleidocranial dysplasia:Etiology, clinicoradiological presentation and management. J Clin and Exp Invest 2012; 3 (1):133-136.
2. Garg RK, Agrawal P. Clinical spectrum of cleidocranial dysplasia: a case report. Cases Journal 2008; 1:1-4.
3. Farronato G; Maspero C, Farronato D, Gioventu S. Orthodontic treatment in a patient with cleidocranial dysostosis. Angle Orthod 2009; 79:178-185.
4. James M J, Jarvinen E, Wang XP, Thesleff I. Different roles of Runx2 during early neural crest-derived bone and tooth development. J Bone Miner res 2006; 21(7):1034-1044.
5. Bei M, Maas R. FGFs and BMP4 induce both Msx1-independent and Msx1-independent signalling pathways in early tooth development. Development 1998; 125:4325-4333.
6. Amresh Reddy P. Cleidocranial dysplasia. A case report and review of literature. NMJ 2012; 1(1):35-37.
7. Mundlos S. Cleidocranial dysplasia:clinical and molecular genetics. J Med Genet 1999; 36:177-182.
8. Londono RM, Lee B: University of Washington, Seattle. GeneReviews™ [Internet]. [Cited 2006 Jan 3].
9. Dixit R, Dixit K, Paramez AR. Cleidocranial dysplasia. Lung India 2010; 27:176-177.
10. Jensen BL, Kreiborg S. Development of the dentition in cleidocranial dysplasia. J Oral Pathol Med 1990; 19:89-93.
11. Furuuchi T, Kochi S, Sasano T, Iikubo M, Komai S, Igari K. Morphologic characteristics of masseter muscle in cleidocranial dysplasia: A report of 3 cases. Oral Surg Oral Med Oral Pathol Oral Radiol Endod 2005; 99:185-90.
12. Pusey RF, Durie JF. A case of cleidocranial dysostosis showing failure of eruption of teeth. Br Dent J 1943; 75 (1):11-13.
| How to Cite This Article: Patil PP, Barpande SR, Bhavthankar JD, Humbe JG. Cleidocranial Dysplasia: A Clinico-radiographic Spectrum with Differential Diagnosis. Journal of Orthopaedic Case Reports 2015 April-June;5(2):21-24. Available from: https://www.jocr.co.in/wp/2015/04/20/2250-0685-264-fulltext/ |

[Full Text HTML] [Full Text PDF] [XML]
[rate_this_page]
Dear Reader, We are very excited about New Features in JOCR. Please do let us know what you think by Clicking on the Sliding “Feedback Form” button on the <<< left of the page or sending a mail to us at editor.jocr@gmail.com