[box type=”bio”] Learning Points for this Article: [/box]
Orthopedic deformities that are found in Hajdu-Cheney syndrome may be challenging to treat and the underlying bone and connective tissue abnormalities that are associated with this disease can complicate operative management.
Case Report | Volume 7 | Issue 5 | JOCR September – October 2017 | Page 11-15| Ashish B. Shah, Breann K Tisano, Osama Elattar, Jackson Rucker Staggers, Sameer Naranje. DOI: 10.13107/jocr.2250-0685.876
Authors: Ashish B Shah [1], Breann K Tisano [1], Osama Elattar [1], Jackson Rucker Staggers [1], Sameer Naranje [1]
[1] Department of Surgery, Division of Orthopaedic Surgery, University Of Alabama at Birmingham, Birmingham, Alabama, USA.
Address of Correspondence
Dr. Ashish B. Shah,
Department of Surgery, Division of Orthopaedic Surgery, University Of Alabama at Birmingham, Birmingham 35205, Alabama, US.
E-mail: ashishshah@uabmc.edu
Abstract
Introduction: Hajdu-Cheney syndrome (HCS) is a rare autosomal dominant disease characterized by acroosteolysis, wormian skull bones with persistent skull sutures, premature loss of teeth, micrognathia, short stature, hypermobility of the joints, neurologic manifestations such as basilar invagination with subsequent paresthesia, hearing loss, and speech alterations, and osteoporosis with tendency to pathologic fractures of long bones and vertebrae as well as painful hands and feet. Very few cases have been earlier reported in the literature.
Case Report: We report a case of a 50-year-old female with bilateral foot deformities as a manifestation of the rare genetic disorder HCS. Surgical management of the left foot consisted of Morton’s neuroma excision and Weil osteotomy with proximal interphalangeal joint resection and Kirschner wire fixation of the second and third metatarsophalangeal (MTP) joints. Recurrent subluxation of the left second MTP joint was observed at 5-week follow-up. The right foot was treated similarly 7 weeks after the initial operation. The post-operative course of the right foot was complicated by bone resorption and nonunion of the second and third metatarsal Weil osteotomies.
Conclusion: Management of complex foot deformities associated with HCS can be challenging and have not previously been described in the literature. Underlying bone and connective tissue abnormalities intrinsic to the syndrome may increase the risk of recurrence after surgical correction. Consideration should be given to such post-operative complications when treating foot deformities in a patient with HCS.
Keywords: Acroosteolysis, basilar invagination, Hajdu-Cheney syndrome, osteoporosis, wormian skull bones.
Introduction
Hajdu-Cheney syndrome (HCS) is a genetic bone disease first described by Hajdu and Kauntze in 1948 [1] and later by Cheney in 1965 [2]. Various terms have been used to describe this syndrome, including the acroosteolysis syndrome, arthrodentoosteodysplasia, hereditary osteodysplasia with acroosteolysis, cranioskeletal dysplasia with acroosteolysis, familial osteodysplasia, and HCS [1, 3, 4, 5]. The etiology is unknown, though a collagen abnormality with a subsequent defect in bone formation has been suggested [2, 4]. The syndrome presents at birth with distinctive dysmorphic features, which become more obvious with advancing age. The characteristic facies, including frontal bossing, mid-facial flattening, thick coarse hair, and low-set ears with enlarged ear lobules, should raise suspicion. Once suspected the characteristic hand findings facilitate the diagnosis [1, 2, 3, 6]. The association of acroosteolysis with any three other distinctive features, such as wormian bones, persistent skull sutures (particularly the lambdoid), micrognathia, platybasia, mid-facial flattening, premature loss of teeth, or short stature, is sufficient to make a diagnosis. In adults, acroosteolysis with a positive family history of HCS is enough for diagnosis [5]. Other less distinctive but supportive features include joint hypermobility, hypoplastic alveolar arches with unerupted teeth, thick coarse hair, hypoplastic mandible and maxilla, a widened sella turcica, basilar invagination with its complications, kyphoscoliosis, and genu valgum [2, 7, 8]. Acroosteolysis of the digits is a defining manifestation of HCS. Very few reports in the literature, however, detail the orthopedic foot pathology [9, 10, 11, 12] and none specifically document their management. The purpose of this paper is to report this rare diagnosis of HCS, with emphasis on the difficulties in the operative management of associated foot deformities.
Case Report
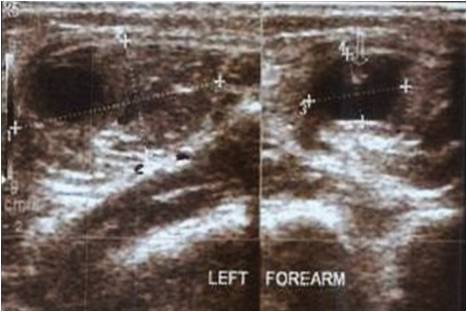
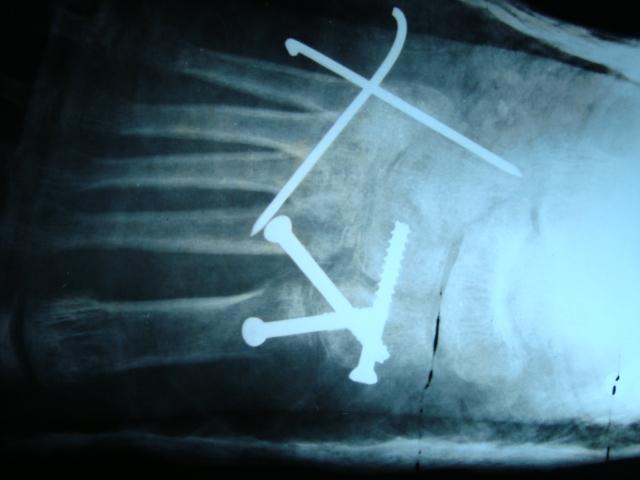
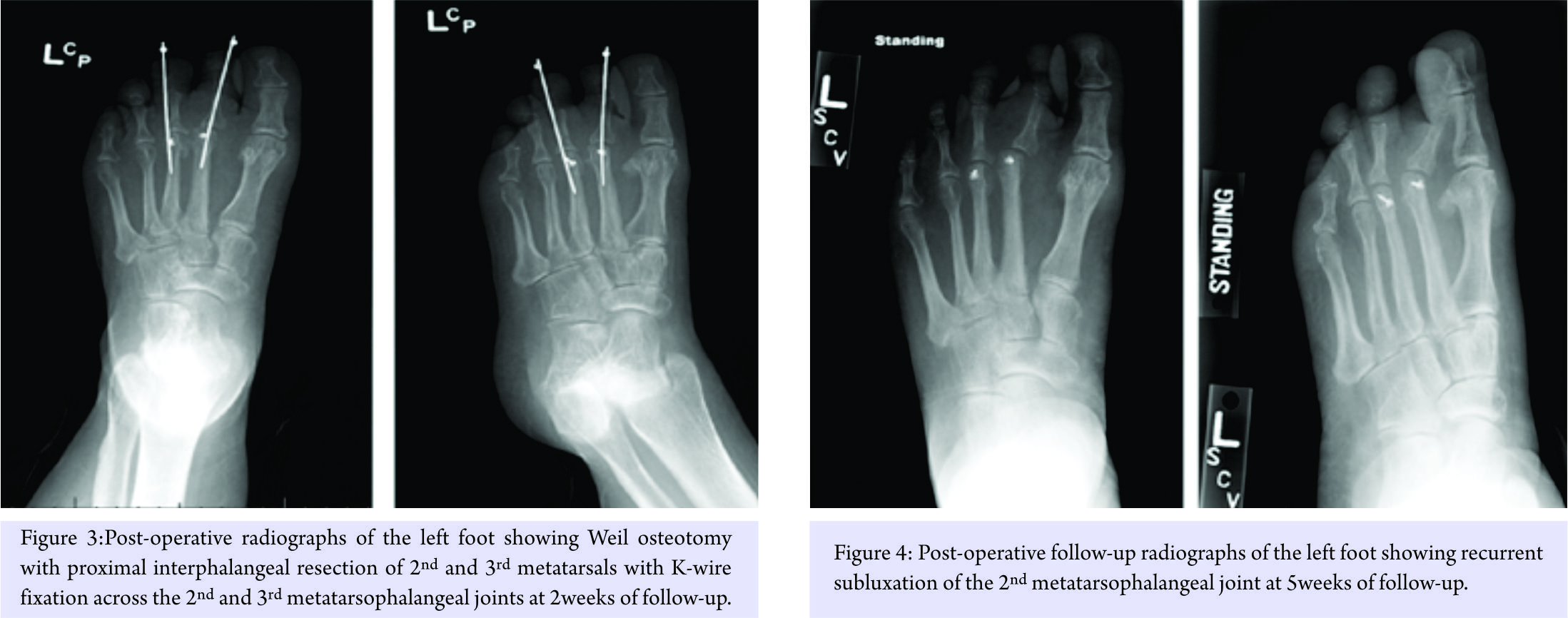
A 50-year-old female patient was referred to the foot and ankle clinic with the existing diagnosis of HCS. She complained of bilateral foot pain and deformities of the toes. Physical examination revealed left-sided dislocation of the second metatarsophalangeal (MTP) joint, subluxation of the third MTP joint, and Morton’s neuroma between the third and fourth toes (Fig. 1). Examination of the right foot similarly demonstrated second MTP joint dislocation and third MTP joint subluxation, along with second-third and third-fourth intermetatarsal Morton’s neuromas (Fig. 2).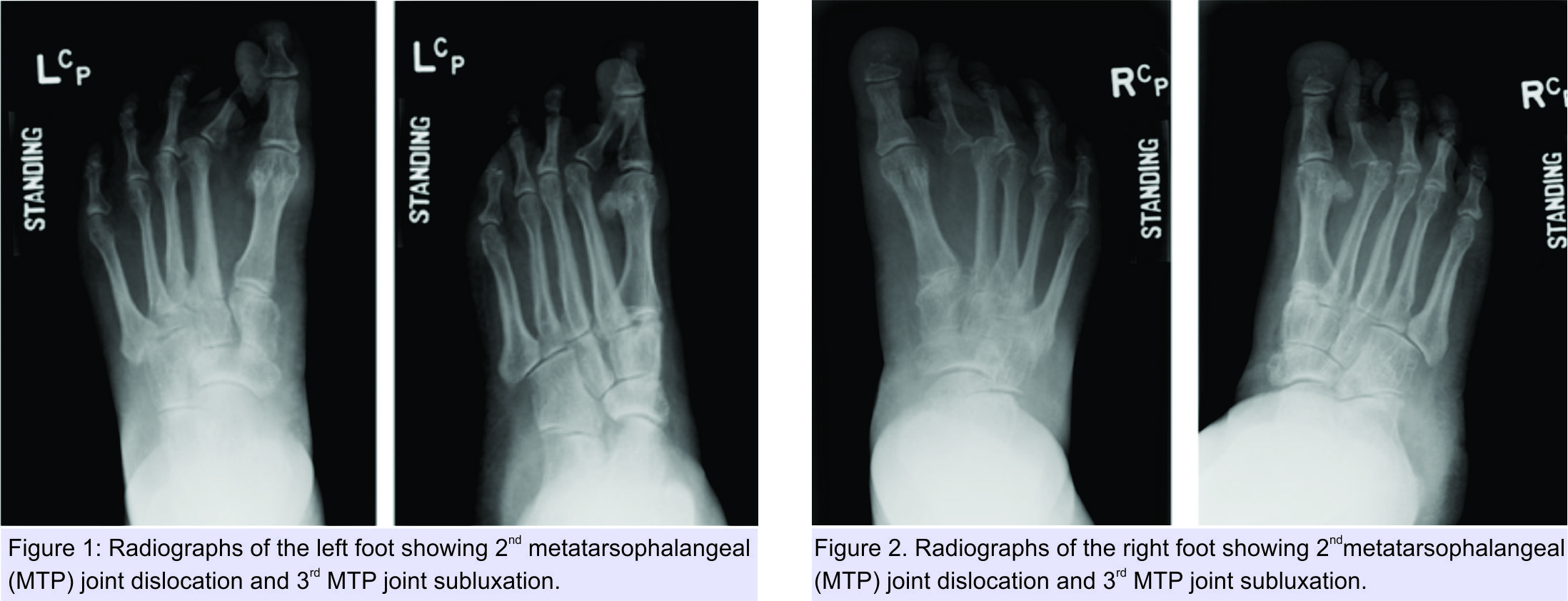 Treatment options were discussed with the patient, including surgical intervention for her foot problems. She was counseled about the high risk of post-operative recurrence of deformities, nonunion, wound complications, and worsening of pain (in setting of pre-existing neuropathy) because of the underlying syndrome. The patient was operated on first on the left foot for Morton’s neuroma excision and Weil osteotomy with proximal interphalangeal (PIP) joint resection of the second and third metatarsals with K-wire fixation across the second and third MTP joints (Fig. 3). Her post-operative course was complicated by recurrent subluxation of the second MTP joint (Fig. 4). A revision surgical option was discussed but refused by the patient, as she was satisfied with the surgery in terms of good pain relief and functional improvement.
Treatment options were discussed with the patient, including surgical intervention for her foot problems. She was counseled about the high risk of post-operative recurrence of deformities, nonunion, wound complications, and worsening of pain (in setting of pre-existing neuropathy) because of the underlying syndrome. The patient was operated on first on the left foot for Morton’s neuroma excision and Weil osteotomy with proximal interphalangeal (PIP) joint resection of the second and third metatarsals with K-wire fixation across the second and third MTP joints (Fig. 3). Her post-operative course was complicated by recurrent subluxation of the second MTP joint (Fig. 4). A revision surgical option was discussed but refused by the patient, as she was satisfied with the surgery in terms of good pain relief and functional improvement.
Following 7-week post-operative recovery period from the left foot surgery, the patient underwent Morton’s neuroma excision and Weil osteotomy with second and third PIP resection and K-wire fixation in the right foot (Fig. 5). Extensor tendon lengthening was also performed in the same foot. Post-operative course was complicated by atrophic nonunion of the osteotomies (Fig. 6), as well as worsening of her neuropathic pain.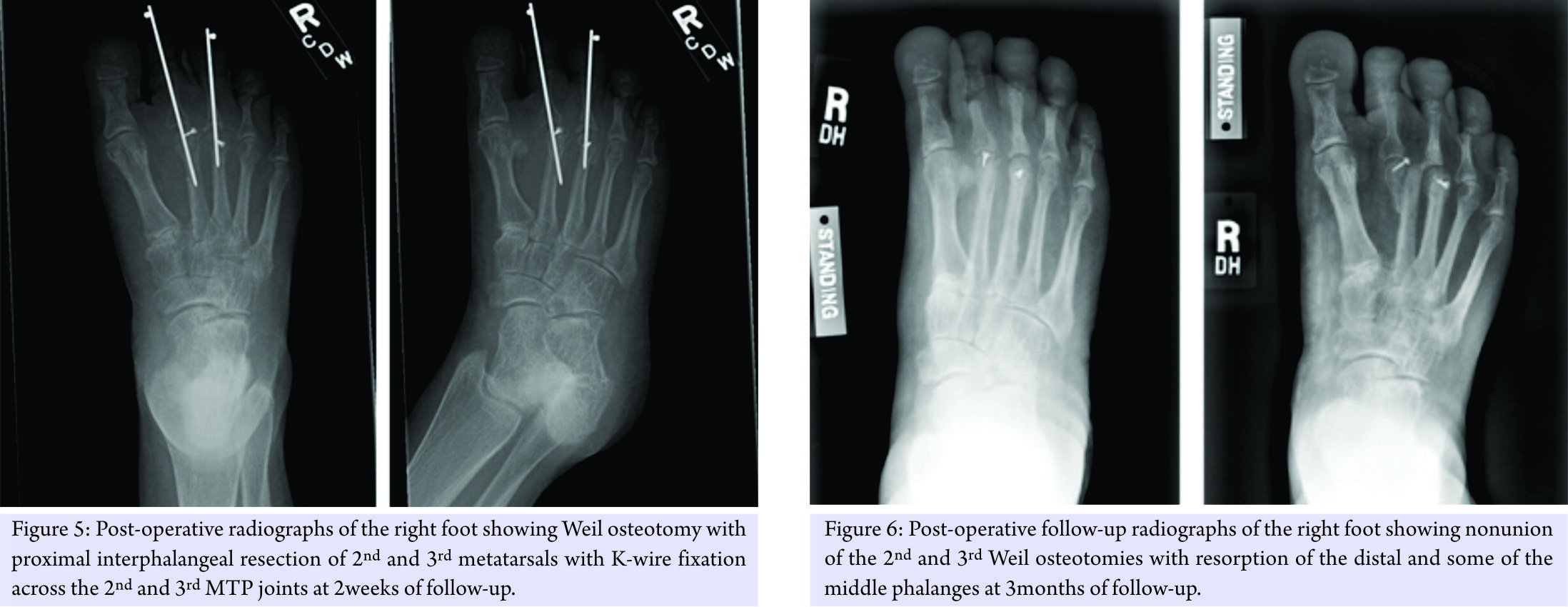
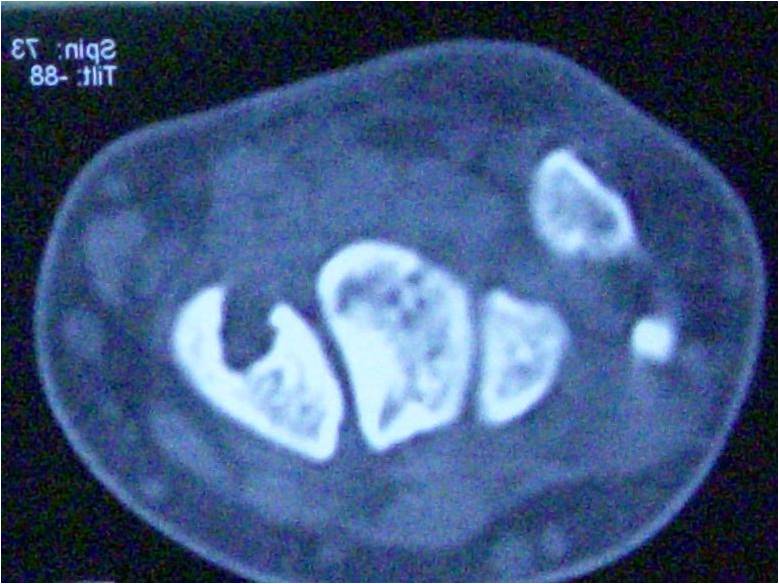
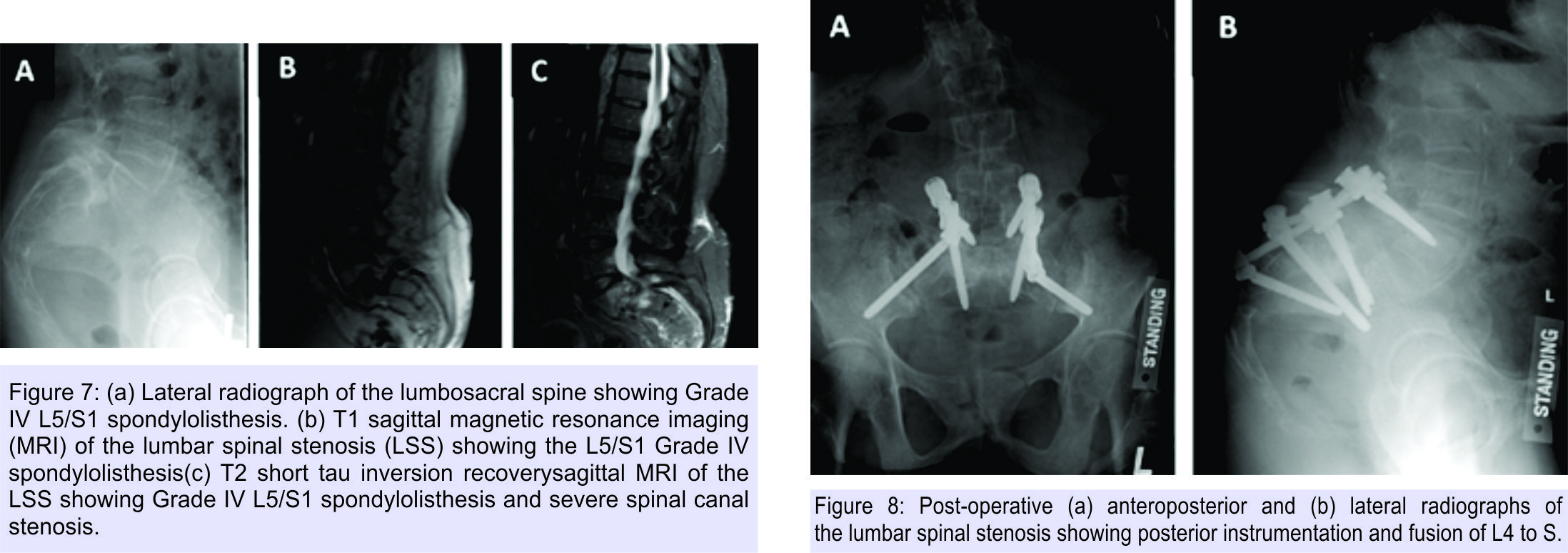
Additional orthopedic manifestations in this patient affected the spine in the form of Grade IV L5/S1 isthmic spondylolisthesis with stenosis and radiculopathy, in the setting of severe osteoporosis (Fig. 7).
After a failed 6-month trial of osteoporosis treatment, she was operated on by our spine colleagues who performed posterior laminectomy at L4-L5 and L5-S1 and posterior instrumentation and fusion, L4 to the sacrum with iliac crest bone graft (Fig. 8).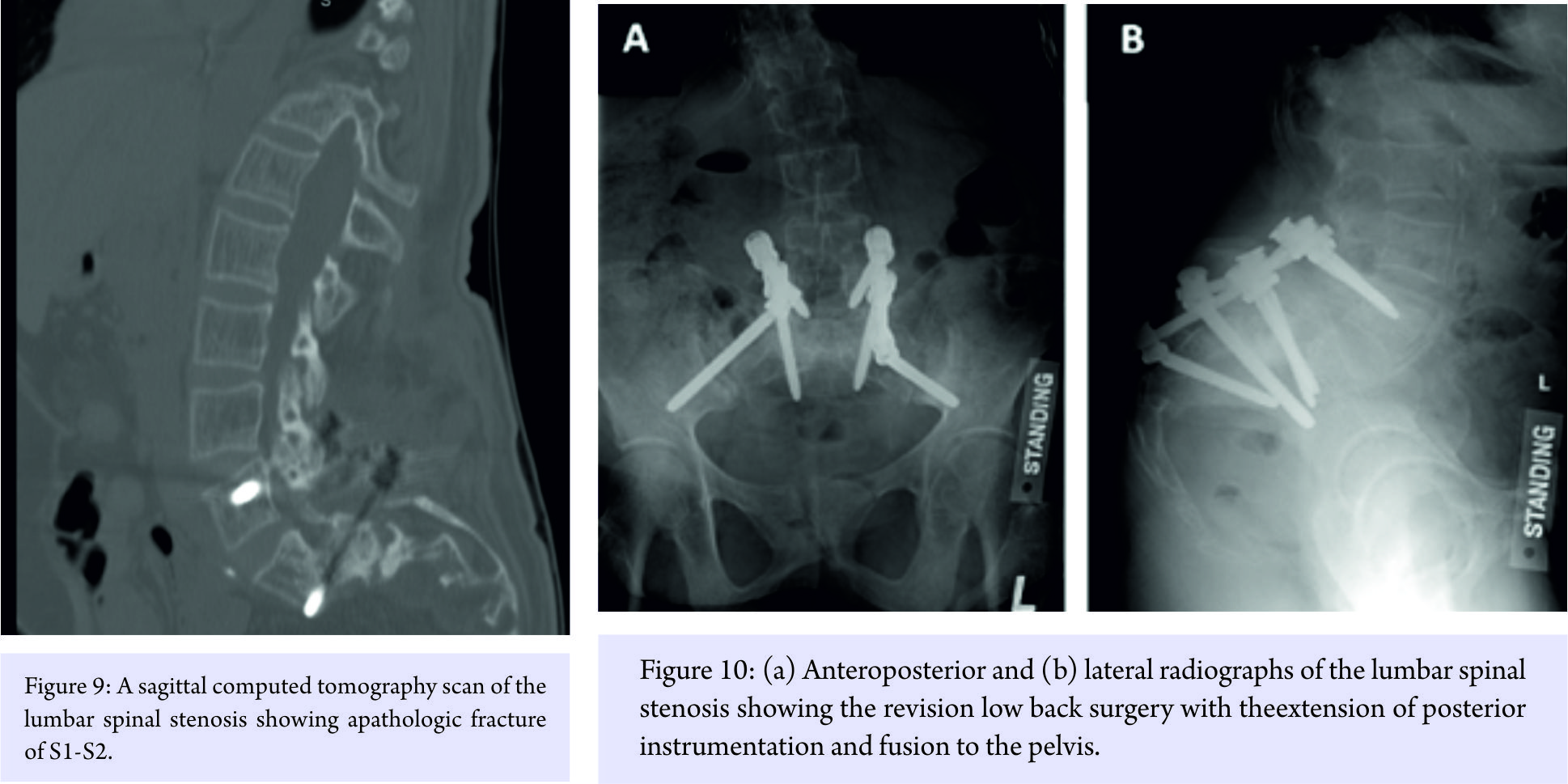 She later developed a pathologic (osteoporotic) fracture S1-S2 (Fig. 9) for which posterior instrumentation was revised with the extension of fusion to the pelvis (Fig. 10).
She later developed a pathologic (osteoporotic) fracture S1-S2 (Fig. 9) for which posterior instrumentation was revised with the extension of fusion to the pelvis (Fig. 10).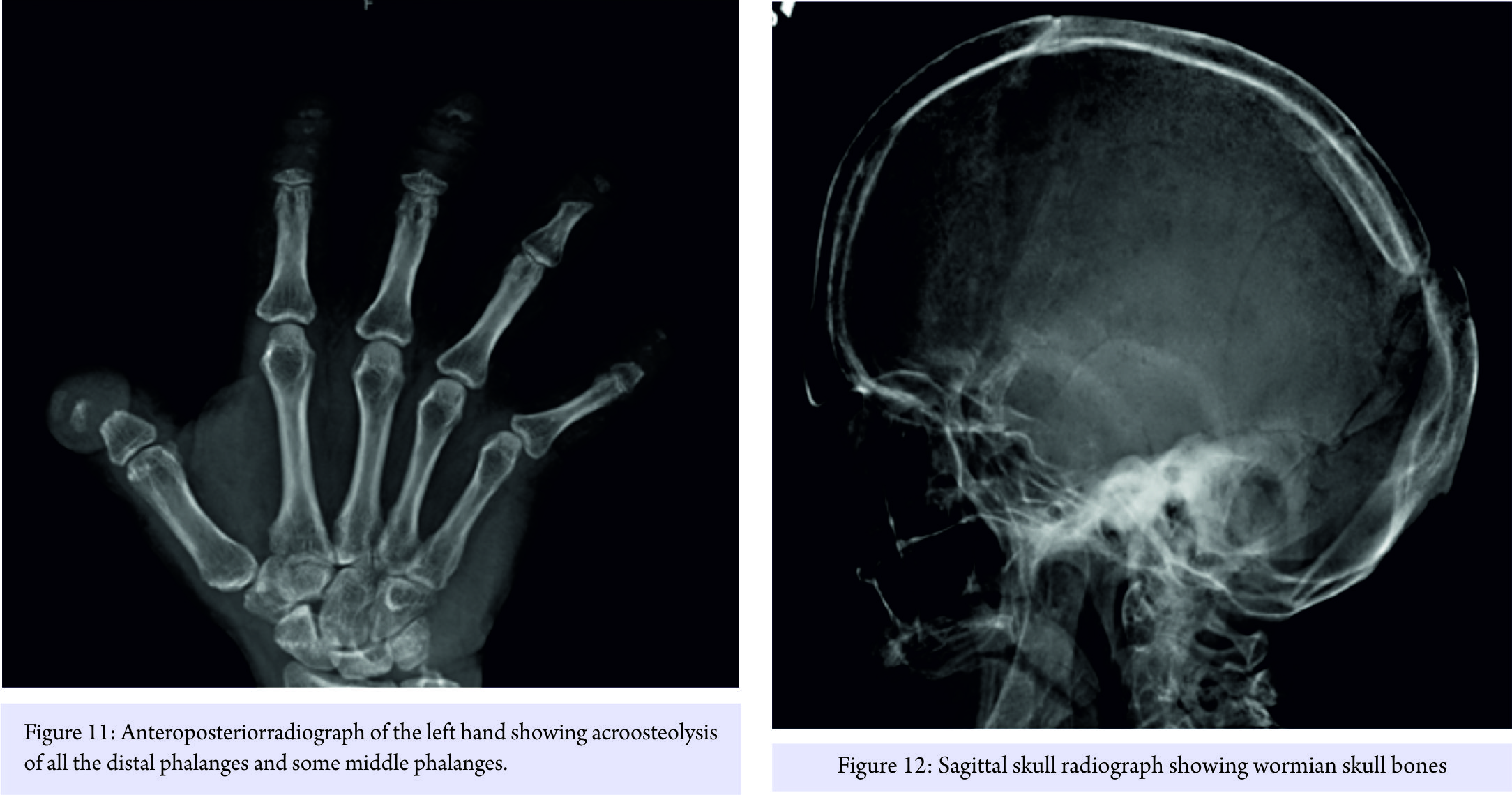 As a point of interest, the following additional manifestations of the HCS were also observed in the patient: Coarse dysmorphic facial features (depression in the coronal region, epicanthic folds, flat nasal bridge, bushy eyebrows, down-slanted palpebrae, malar hypoplasia, relative prognathism, low-set and posteriorly rotated ears, and loss of teeth), coarse thick hair, acroosteolysis of all distal and some middle phalanges of the hand (Fig. 11), wormian skull bones (Fig. 12), history of surgical correction of patent ductus arteriosus, osteoporosis, scoliosis (Fig. 13), platybasia, basilar invagination of skull (Fig. 14), hearing difficulty, neuropathy, umbilical hernia, and esophageal stricture with cervical web (treated with endoscopic dilatation).
As a point of interest, the following additional manifestations of the HCS were also observed in the patient: Coarse dysmorphic facial features (depression in the coronal region, epicanthic folds, flat nasal bridge, bushy eyebrows, down-slanted palpebrae, malar hypoplasia, relative prognathism, low-set and posteriorly rotated ears, and loss of teeth), coarse thick hair, acroosteolysis of all distal and some middle phalanges of the hand (Fig. 11), wormian skull bones (Fig. 12), history of surgical correction of patent ductus arteriosus, osteoporosis, scoliosis (Fig. 13), platybasia, basilar invagination of skull (Fig. 14), hearing difficulty, neuropathy, umbilical hernia, and esophageal stricture with cervical web (treated with endoscopic dilatation).
Discussion
HCS is a rare autosomal dominant disorder characterized principally by acroosteolysis (dissolution) of the distal phalanges with possible associated digit abnormalities, distinctive craniofacial and skull features, dental anomalies, and proportionate short stature. While dysmorphic features at birth and presence of acroosteolysis allow diagnosis early in life, the full phenotype is rarely present in childhood. Instead, the various clinical and radiologic abnormalities manifest at different ages and often progress with time [1, 3]. Treatment of this disorder is generally symptomatic and the prognosis is quite good. Morbidity is mostly related to bone changes secondary to acroosteolysis as well as basilar invagination resulting in neurological complications. Presentation and management of toe and foot deformities in patients with HCS are not well described in the literature. Although previous reports have described the management of other manifestations of HCS [6, 7, 13, 14], we were unable to find any specifically addressing management of foot deformities in these patients. Acroosteolysis and underlying collagen abnormalities intrinsic to HCS predispose these patients to the poor post-operative bone and soft tissue healing. There are few reports describing orthopedic challenges and complications associated with operative management in these patients [15, 16]. Anecdotally, any surgical intervention done for bony and joint deformities may thus have a higher complication rate of nonunion and recurrence. Our patient was noted at 5-week post-operative follow-up to have a recurrence of the deformity of the left second MTP joint after removal of K-wire fixation. Failure of symptomatic relief may be affected by progressive neuropathy [17], which was also observed in our patient. As such, any bony or soft tissue surgical intervention should be sought with caution and consideration of these post-operative complications.
Conclusion
HCS is a rare disorder associated with complex foot pathology. Underlying bone and connective tissue abnormalities may affect the post-operative clinical outcome if surgical intervention is performed. This case report underscores the difficulty in management of these foot deformities and the likelihood of recurrence.
Clinicla Message
HCS is a rare condition with a diverse constellation of skeletal manifestations. Deformities in the foot have the propensity to recur after surgical management. It is important for physicians to consider underlying bone and connective tissue abnormalities and their effect on post-operative clinical outcome.
References
1. Hajdu N, Kauntze R. Cranio-skeletal dysplasia. Br J Radiol 1948;21:42-8.1. Hajdu N, Kauntze R. Cranio-skeletal dysplasia. Br J Radiol 1948;21:42-8.
2. Cheney W. Acro-osteolysis. Am J Roentgenol Radium Ther Nucl Med 1965;94:595-607.
3. Brennan A, Pauli RM. Hajdu-cheney syndrome: Evolution of phenotype and clinical problems. Am J Med Genet 2001;100:292-310.
4. Brown D, Bradford DS, Gorlin RJ, Desnick RJ, Langer LO, Jowsey J, et al. The acro-osteolysis syndrome: Morphologic and biochemical studies. J Pediatr 1976;88:573-80.
5. Zugibe F, Herrmann J, Opitz JM, Gilbert EF, McMillian G. Arthrodentoosteodysplasia: A genetic “Acroosteolysis” syndrome. Birth Defects Orig Artic Ser 1974;10:145-52.
6. Van den Houten B, Ten Kate LP, Gerding JC. The Hajdu-Cheney Syndrome, a review of the literature and report of 3 cases. Int J Oral Surg 1985;14:113-25.
7. Adès LC, Morris LL, Haan EA. Hydrocephalus in Hajdu-Cheney syndrome. J Med Genet 1993;30:175.
8. Marik I, Kuklik M, Zemkowa D, Kozlowski K. Hajdu-Cheney syndrome: Report of a family and a short literature review. Aust Radiol 2006;50:534-8.
9. Palav S, Vernekar J, Pereira S, Desai A. Hajdu-Cheney syndrome: A case report with review of literature. J Radiol Case Rep 2014;8:1-8.
10. Letchumanan P, Thumboo J, Leong RT. A patient with progressive shortening of the fingers. J Rheumatol 2009;36:198-9.
11. Sahin A, Pepeler MS, Shimbori N. A patient with acro-osteolysis syndrome: Hajdu-Cheney. Intern Med 2010;49:87-8.
12. Gorlin RJ, Cohen MM, Hennekam RCM, eds. Syndromes of the Head and Neck, 4th ed. Oxford University Press, New York, NY. 2001:315-318
13. Diren H, Kovanlikaya I, Suller A, Dicle O. The Hajdu-Cheney syndrome: A case report and review of the literature. Pediatr Radiol 1990;20:568-9.
14. Weleber RG, Beals RK. The Hajdu-Cheney syndrome. Report of two cases and review of the literature. J Pediatr 1976;88:243-9.
15. Mattei TA, Rehman AA, Issawi A, Fassett DR. Surgical challenges in the management of cervical kyphotic deformity in patients with severe osteoporosis: An illustrative case of a patient with Hajdu Cheney syndrome. Eur Spine J 2015;24:2746-53.
16. Fujioka H, Tanaka J, Yoshiya S, Kokubu T, Fujita K, Makino T, et al. Proximal translation of the radius following arthroplasty of the distal radioulnar joint in Hajdu-Cheney syndrome. J Shoulder Elbow Surg 2003;12:97-100.
17. Connor A, Highton J, Hung NA, Dunbar J, MacGinley R, Walker R. Multicentric carpal-tarsal osteolysis with nephropathy treated successfully with cyclosporine A: A case report and literature review. Am J Kidney Dis 2007;50:649-54.
 |
 |
 |
 |
 |
| Dr. Ashish B Shah | Dr. Breann K Tisano | Dr. Osama Elattar | Dr. Jackson Rucker Staggers | Dr. Sameer Naranje |
| How to Cite This Article: Shah A, Tisano BK, Elattar O, Staggers JR, Naranje S. Foot Deformities in Hajdu-Cheney Syndrome: A Rare Case Report and Review of the Literature. Journal of Orthopaedic Case Reports 2017 Sep-Oct ; 7(5): 11-15 |
[Full Text HTML] [Full Text PDF] [XML]
[rate_this_page]
Dear Reader, We are very excited about New Features in JOCR. Please do let us know what you think by Clicking on the Sliding “Feedback Form” button on the <<< left of the page or sending a mail to us at editor.jocr@gmail.com