[box type=”bio”] Learning Point of the Article: [/box]
Giant cell tumors of soft tissues should be diagnosed and treated appropriately to avoid reoccurrence.
Case Report | Volume 9 | Issue 1 | JOCR January – February 2019 | Page 70-73 | Doreen Mensah, Kishor Chhantyal, Yun Xiang Lu, Zhi Yong Li. DOI: 10.13107/jocr.2250-0685.1314
Authors:
[1]Department of Orthopedic Trauma and Joint Surgery, Hospital of Sun-Yat-Sen University, Guangzhou 510010, Guangdong, China,
[2]Department of Spine Surgery, Hospital of Sun-Yat-Sen University, Guangzhou 510010, Guangdong, China.
Address of Correspondence:
Dr. Zhiyong Li,
Department of Orthopedic Trauma and Joint Surgery, Hospital of Sun-Yat-Sen University, Guangzhou 510010, Guangdong, China.
E-mail: doclizhiyong8369@163.com
Abstract
Introduction: Giant cell tumor of soft tissue (GCT-ST)is a low-grade ST tumor that occurs most frequently below the skin and sometimes, extending deeper into the tissues. The tumor is usually hard, painless and considered rare in ST.
Case Report: We report a rare case of a middle-aged woman that presented with a tumor arising from the right tensor fasciae latae muscle. The patient initially opted for traditional Chinese herbal treatment, which we believe, aggravated the growth of the tumor. Radiographic imaging showed clear tumor margins. The histopathological biopsy of the lesion located in the tensor fasciae latae muscle showed a mixture of mononucleated and multinuclear giant cells composed of mesenchyme spindle-shaped cells positive for CD34 and CD88. The patient was treated through surgical resection of the tumor and scheduled for close follow-up.
Conclusion: Since GCT-ST is rare, they are often initially misdiagnosed. These tumors have the tendency to increase in size exponentially within a short period of time, which is why clinical presentation and imaging are necessary for a better pre-operative diagnosis. Radical surgical excision or radiotherapy with close follow-up is recommended for all cases due to the high rate of reoccurrence.
Keywords: Giant cell tumor, tensorfasciae latae, intramuscular tumor, case report, middle-aged female.
Introduction
In surgical practice, soft tissue (ST) tumors are known to be extremely rare with unpredictable behavior. Salm and Sissons first described giant cell tumors-STs(GCT-STs) in 1972. Such tumors present in different shapes and sizes and can be found in any part of the body with the majority of reported cases appearing in the extremities, with the thigh being the most commonly affected site [1, 9]. Clinically, GCT-STs manifest as an asymptomatic mass, but some of these tumors are very aggressive and have the potential to increase in size within a short period of time [2]. As a result of this aggressive behavior, they often metastasize to surrounding connective and adipose tissues, skeletal muscles, and blood and lymph vessels[3].The vast majority of these tumors are successfully treated with clean surgical excision, while others may require additional treatment regimens. Lipoma, hemangioma, and giant cell carcinomas are among the most commonly seen ST tumors, radiographic imaging and tissue biopsies are the best diagnostic techniques in determining the marginal location and the type of tumor, respectively. In this paper, we present the clinical, histologic, and radiologic features of a case of a malignant GCT-ST in a 44-year-old female who presented with an extremely painful right hip mass that aggressively progressed over 6months. The tumor has been completely excised with surgery and follow-up has been advised to monitor reoccurrence.
Case Report
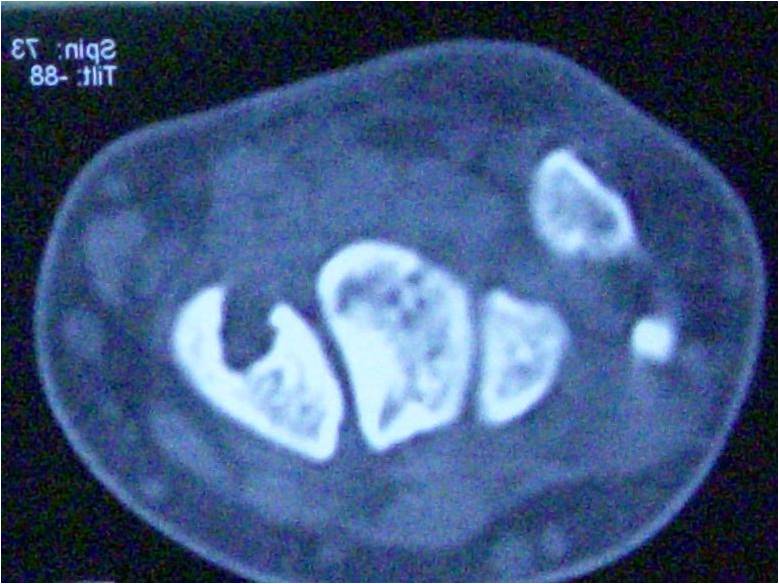
A 44-year-old Asian female presented with an extremely aggressive intramuscular, tender upper right thigh mass of 350mm × 250mm which had progressed over a 6-month duration without any antecedent of infection or trauma. The patient mentioned the size of the tumor was non-remarkable in the first 2 months when she first discovered it in October 2017 but progressed rapidly despite using traditional Chinese medicine and moxibustion. The lesion at the time it was presented to our department on March 2018 had a fungating skin appearance and ST involvement (Fig. 1 and Fig. 2) with a foul-smelling odor. The patient before presentation had used traditional Chinese herbal medicine and moxibustion to treat the tumor, but there was no remarkable improvement after 4 months of treatment. Apart from a previously treated mild thalassemia, there was no other significant preexisting medical history or any known allergies. On examination, the tumor appeared to be tender and soft to touch. Computed tomography and positron emission tomographyscans showed a well-demarcated tumor margin with no bone involvement confined to the tensor fasciae latae muscle (Fig. 3and Fig. 4). After all standard pre-operativeevaluation and preparation, the tumor was successfully resected (Fig. 5 and Fig. 6), followed by additional post-operative medical treatments of pain management, infection prevention, diet modifications, and other conventional rehabilitation procedures.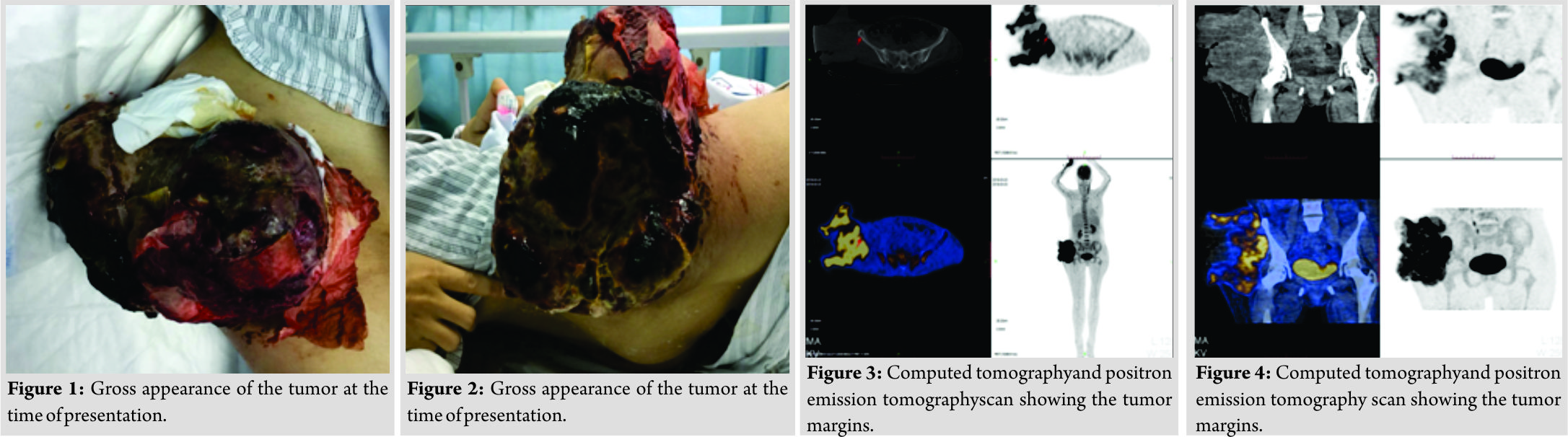
Immunohistochemical study
Microscopic study of the tissue sample revealed that the tumor was located in the intramuscularly with a relatively clear local boundaries, the tumor cells were diffuse and rich in cytoplasm. The nucleus fat showed spindle, oval, round, and polygonal shapes. There was pleomorphic and heteromorphosis mixture of mononucleated and multinuclear giant cells. There was an obvious hemorrhagic necrosis due to the blockage of venous drainage. However, there was the presence of rich small interstitial blood vessels, part of the region seems to have tumorous osteogenesis. In accordance with clinical history, the tumor conforms to giant cell carcinoma of ST.(Fig. 7a and Fig. 7b) Ck(−), Vimt (+), S100(−), smooth muscle actin (SMA) (+), desmin (−), CD34 (blood vessel+), CD88(increase+),ki-67(about 50%+).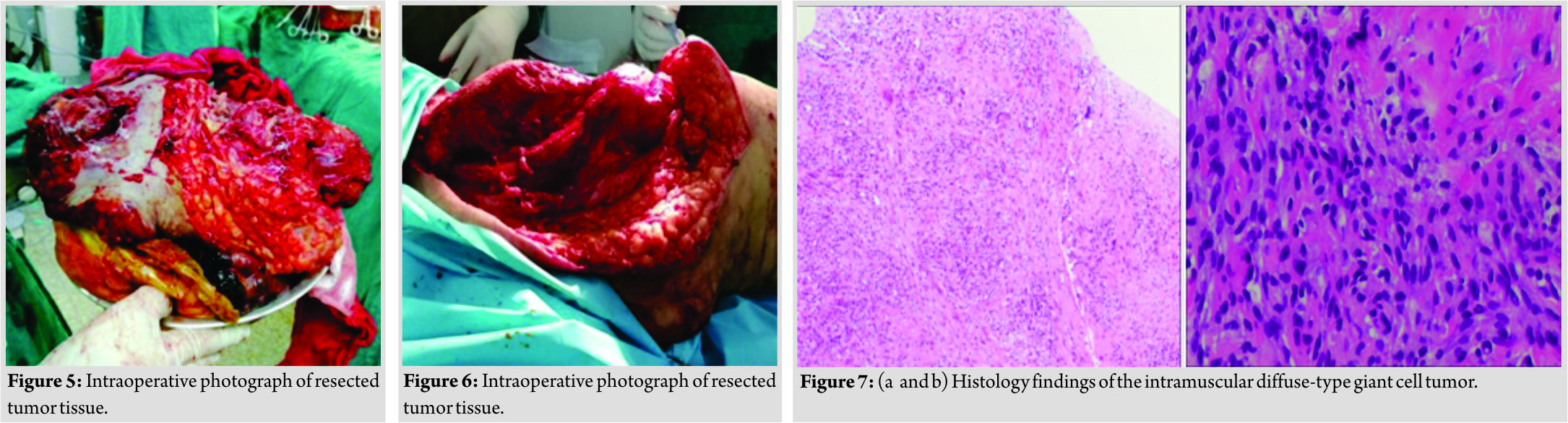
Discussion
The GCT-ST, which is an extremely rare tumor, resembles its osseous variants both histologically and clinically [4,5]. It was first reported as early as 1972 in a case series of 10 benign primary ST tumors [6]. These tumors can exhibit aggressive malignant transformation, abundant mitotic activity, and pleomorphism [6]. However, GCT-ST is predominantly benign in nature, with a high possibility recurrence possibility if inadequately excised. The majority of the cases of GCT-ST occur in the extremities. However, cases occurring in rare locations such as the head, neck, and ears have been reported [7, 8, 11]. Until now, etiological factors for GCT-ST have not been identified, and some reports suggest that chronic inflammation may have a role in its pathogenesis, which remains unproven to date [7]. GCT-ST mostly presents as a painless mass with an average symptom duration of 6 months–1 year [7,12].The mass, in our case, was tender, huge, and with loss of the skin. In our case, microscopic examination showed diffuse tumor cells arranged in a crisscross pattern. Furthermore, there were unclear cell boundaries and multinucleated giant cells. Atypia was obvious. Our immunohistochemistry results indicated vimentin(+), CD68 (high), and SMA negative [8], CD34 (positive for blood vessels), CK(−), S-100(−), desmin (−), and Ki-67. The Ki-67 labeling index was approximately (M ref). The osteoclast-like giant cells usually exhibit positive CD68 and negative CD34 immunostaining [9,10]. GCT-ST is often initially misdiagnosed [4, 11], which was the case of our patient. It was initially diagnosed as a lipoma. These tumors can grow exponentially in a short period of time. Radical surgical excision or radiotherapy with close follow-up is recommended for all cases due to the high rate of reoccurrence. In this case, we opted for radical excision because the tumor was extremely large and adversely affected her daily life activities. Second, due to its size, it had invaded the muscular layer. Conservative surgical resection with tumor-free margin is the typical treatment for GCT-ST that assures a good prognosis with very low recurrence rate[11]. Magnetic resonance imaging (MRI) scans are the most reliable radiographic diagnostic choice for identifying the margins and invaded ST because they are highly sensitive.
Conclusion
We reported a case of intramuscular diffuse-type GCT-ST in 44 years old. The tumor was exceptionally aggressive and local reoccurrence after surgery cannot be predicted. GCT-STs are clinically and morphologically identical to GCT of the bone, which is why they can progress rapidly and aggressively if not properly and promptly treated. Radical and complete surgical excision is the most reliable treatment approach. Reoccurrence is very common; therefore, close monitoring during follow-up is recommended. In the case of this patient, the growth of this tumor was likely due to the delay in surgical treatment in addition to the use of herbal moxibustion, which likely escalated the growth. Due to how rare GCT-STs are, we hope this case serves as an instructive for prompt diagnosis and treatment of any primary intramuscular tumors.
Consent
The patient was asked if the data concerning the case can be used for publication, and they consented.
Clinical Message
Diagnosing GCT-ST can be difficult both radiographically and clinically because they are extremely rare with unpredictable behavior. However, MRI is the most reliable radiographic diagnostic choice for identifying the tumor margins and invaded ST. Radical surgical resection of the tumor is the treatment for GCT-ST that assures a good prognosis. Reoccurrence is very common; therefore, close monitoring of the patient post-operativeis recommended.
References
1. Asotra S, Sharma S. Giant cell tumor of soft tissue. Cytological diagnosis of a case. J Cytol 2009;26:33-5.
2. O’Connell JX, Wehrli BM, Nielsen GP, Rosenberg AE. Giant cell tumors of soft tissue: A clinicopathologic study of 18 benign and malignant tumors. Am J Surg Pathol 2000;24:386-95.
3. Angervall L, Hagmar B, Kindblom LG, Merck C. Malignant giant cell tumor of soft tissues: A clinicopathologic, cytologic, ultrastructural, angiographic, and microangiographic study. Cancer 1981;47:736-47.
4. Mokrani A.Guermazi F, Yahyaoui Y, Hmida L, Doghri R. Giant cell tumor of soft tissues. A case report and review of literature. J Cancer Sci Ther Am 2017;9:562-5.
5. Saldanha P, Tak HN. Giant cell tumour of soft tissue – A rare neoplasm. J Clin Biomed Sci 2014;4:326-9.
6. Bhat A. Soft tissue giant cell tumour of low malignant potential: A rare tumour at a rare site. J Clin Diagn Res 2013;7:2960-1.
7. Hafiz S, Shaheen M, Awadh N, Esheba G. Giant cell tumor of soft tissue. A case report for the first time in ear. Hum Pathol Case Rep Am 2017;10:12-4.
8. Rodríguez-Peralto JL, López-Barea F, Sánchez-Herrera S, Atienza M. Primary aneurysmal cyst of soft tissues (extraosseous aneurysmal cyst) Am J Surg Pathol 1994;18:632-6.
9. Klenke FM, Wenger DE, Inwards CY, Rose PS, Sim FH. Giant cell tumor of bone: Risk factors for recurrence. Clin Orthop Relat Res 2011;469:591-9.
10. Vanni D, Pantalone A, Andreoli E, Caldora P, Salini V. Giant cell tumor of the distal ulna: A case report. J Med Case Rep 2012;6:143.
11. Mazhari NJ, Dhal A, Mandal AK. Giant cell tumour of the soft tissue. A case report. Indian J Pathol Microbiol 2000;43:155-6.
 |
 |
 |
 |
| Dr. Doreen Mensah | Dr. Kishor Chhantyal | Dr. Yun Xiang Lu | Dr. Zhi Yong Li |
| How to Cite This Article: Mensah D, Chhantyal K, Lu Y X, Li Z Y. Malignant Giant Cell Tumor of Soft Tissue: A Case Report of Intramuscular Malignant Giant Cell Carcinoma of the Tensor Fasciae Latae. Journal of Orthopaedic Case Reports 2019 Jan-Feb; 9(1): 70-73. |
[Full Text HTML] [Full Text PDF] [XML]
[rate_this_page]
Dear Reader, We are very excited about New Features in JOCR. Please do let us know what you think by Clicking on the Sliding “Feedback Form” button on the <<< left of the page or sending a mail to us at editor.jocr@gmail.com