[box type=”bio”] Learning Point of the Article: [/box]
Staged procedures, angioembolization, characterization as secretary versus non-secretary, and a team approach involving the endocrinologists are the keys to success while dealing with neuroendocrine tumors.
Case Report | Volume 9 | Issue 5 | JOCR September – October 2019 | Page 39-42 | Sudhir Srivastava, Aditya Raj, Sunil Bhosale, Nandan Marathe. DOI: 10.13107/jocr.2019.v09i05.1524
Authors: Sudhir Srivastava[1], Aditya Raj[1], Sunil Bhosale[1], Nandan Marathe[1]
[1]Department of Orthopedics, Seth Gordhandas Sunderdas Medical College and King Edward Memorial Hospital, Mumbai, Maharashtra, India.
Address of Correspondence:
Dr. Aditya Raj,
Department of Orthopedics, MS (Ortho), 6th Floor, Multistorey Building, Seth Gordhandas Sunderdas Medical College and King Edward Memorial Hospital, Parel, Mumbai- 400012, Maharashtra, India.
E-mail: adityagmck@gmail.com
Abstract
Introduction: Paragangliomas are relatively rare group of well-vascularized tumor having an origin from the embryonic neural crest cells. They arise from extra-adrenal autonomic paraganglia and have neuroendocrine cells that can secrete catecholamines. Most head and neck paragangliomas are parasympathetic type and are non-functional. They are located near the skull base and along the vagus nerve. Paragangliomas of the thoracoabdominal regions are sympathetic type and known to secrete catecholamines. It is an important cause for episodic hypertension. Triad of episodic hypertension, sweating, and palpitations is included in the classical triad for the catecholamine secreting tumors. Rarely, the paraganglioma can show features of malignancy in 10–15% of cases.
Case Report: We present here a 34-year-old female who came to us with back pain, episodic hypertension associated with palpitations, and sweating. Back pain was not associated with other neurological symptoms and neuroclaudication. She was treated with en bloc resection of the tumor with L1 corpectomy posterior instrumentation and anterior reconstruction.
Conclusion: Secretory paravertebral paraganglioma of the lumbar spine is infrequently reported. Its management requires multidisciplinary approach with optimization of perioperative blood pressure and meticulous handling of the tumor mass. In cases, where the vertebral body is destroyed, it requires anterior column reconstruction for stabilization.
Keywords: Paraspinal, malignant, secretory, paraganglioma, lumbar vertebra.
Introduction
Paragangliomas are relatively rare group of well-vascularized tumor having an origin from the embryonic neural crest cells. They arise from extra-adrenal autonomic paraganglia and have neuroendocrine cells that can secrete catecholamines. Most head and neck paragangliomas are parasympathetic type and are non-functional. They are located near the skull base and along the vagus nerve. Paragangliomas of the thoracoabdominal regions are sympathetic type and known to secrete catecholamines. It is an important cause for episodic hypertension[1]. Triad of episodic hypertension, sweating, and palpitations is included in the classical triad for the catecholamine secreting tumors. Rarely, the paraganglioma can show features of malignancy in 10–15% of cases. We present here a 34-year-old female who came to us with back pain, episodic hypertension associated with palpitations, and sweating. Back pain was not associated with other neurological symptoms and neuroclaudication. She was treated with en bloc resection of the tumor with L1 corpectomy posterior instrumentation and anterior reconstruction. Secretory paravertebral paraganglioma of the lumbar spine is infrequently reported. Its management requires multidisciplinary approach with optimization of perioperative blood pressure and meticulous handling of the tumor mass. In cases, where the vertebral body is destroyed, it requires anterior column reconstruction for stabilization.
Case Report
A 34-year-old female presented with dull low back pain radiating to bilateral lower limb and paresthesias in the lower limb of 2 years duration. It was associated with episodic palpitation, perspiration, and occasional headache for 2 years. Medical history was positive for two episodes of hypertensive urgency which required inpatient admission and management. However, at no time, further evaluation was carried out to investigate the etiology for the hypertension. On general examination, she was a female in her early 30s averagely build and nourished. Vitals were within normal limits with the exception of few episodes of hypertension where the BP reached maximum of 220/140 mmHg. Low back pain was not associated with any spinal tenderness and neurological examination was unremarkable. Laboratory investigations revealed that plasma-free normetanephrine was 4555pg/ml (Normal= 0–196). Plain radiographs of the lumbosacral spine showed scalloping of the anterior border of L1 vertebra and degenerative changes lower down at L4-L5 (Fig. 1). Magnetic resonance imaging (MRI) of the lumbar spine revealed heterogeneously enhancing retroperitoneal lesion at L1 with areas of necrosis (Fig. 2-4). Both adrenal glands were unremarkable. Computed tomography (CT) abdomen and pelvis were suggestive of hyperenhancing lobulated mass measuring 29× 49 mm axially closely abutting abdominal aorta for more than 90° but <180° with loss of intervening fat planes at L1 with scalloping and anterior destruction of L1 vertebral body. Positronemission tomography (PET) scan was suggestive of metabolically active lesion seen at L1 and L2 vertebra (Fig. 5). Scintigraphic evidence of metaiodobenzylguanidine (MIBG) avid mass in upper abdomen suggesting paraganglioma was found on iodine-123-MIBG scan (Fig. 6). The patient required pre-operative optimization of blood pressure with the combination of alpha-blockers and beta-blockers in optimum dosage before surgical resection. The patient underwent angioembolization before the surgery in view of high vascularity to reduce the intraoperative blood loss. Bilateral feeders from lumbar arteries were embolized with 100–200µ particles. Surgical approach was planned as a staged procedure. In the first stage in prone position under general anesthesia, a midline posterior approach was used. Pedicular screw instrumentation (except at L1) and fusion were performed from D11 to L3 (Fig. 7). In the second stage, the patient underwent excision of tumor mass with L1 corpectomy and anterior reconstruction with an expandable titanium cage and tricortical onlay graft by a retroperitoneal approach in the right lateral position (Fig. 8-10). The graft was harvested from the left iliac crest. Intraoperatively, there were fluctuations of blood pressure during tumor removal which was handled well with inotropic support. Histopathology report was suggestive of cells in nests, “Zellballen”(ball of cells) pattern. On immunohistochemistry, tumor cells were positive for chromogranin A, S-100, Cyclin-D1, and weakly positive for synaptophysin. Nuclei showed atypia; however, mitotic activity was absent. The patient was immobilized for 3 weeks in bed following which she was mobilized with a Taylor’s brace. The patient remained neurologically intact following the second stage of surgery and is at present asymptomatic 8 months following the surgery with no complaints of backache, palpitations, or hypertension.
Discussion
The 2004 WHO classification of endocrine tumors has classified paraganglioma to be intra-adrenal or extra-adrenal. Intra-adrenal paragangliomas are catecholamines secreting and are also known as pheochromocytomas arising from the adrenal medulla.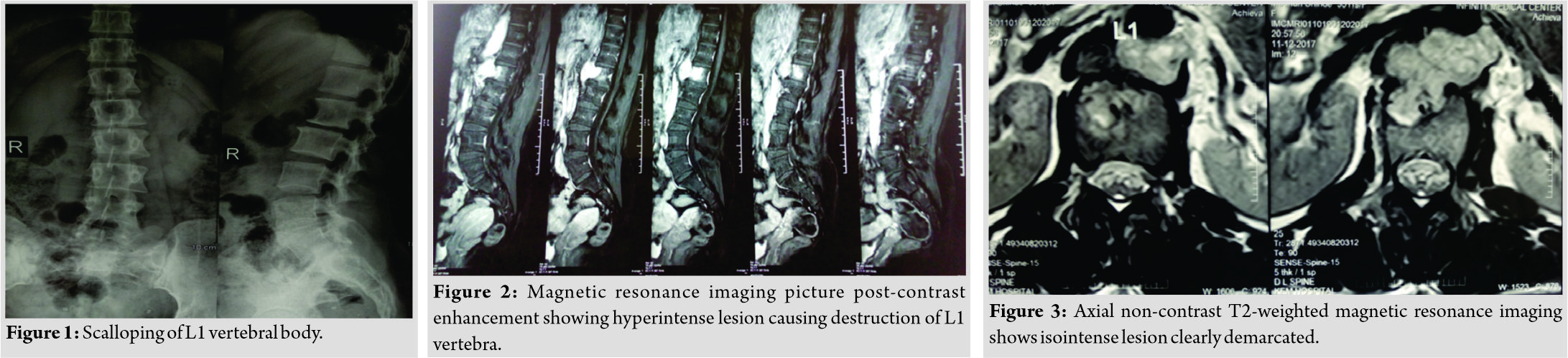
All extra-adrenal tumors originating from sympathetic and parasympathetic ganglia irrespective of secretory status are termed “extra-adrenal paraganglioma”[1]. Presence of typical histological features of mitoses, cellular atypia and vascular invasion is usually suggestive of malignancy, but in these tumors it cannot reliably differentiate tumors which metastasize from the ones which do not [2]. Young age, family history, and presence of malignancy should compel to work up for genetic screening of the patients. Two-thirds of extra-adrenal tumors can be associated with familial syndromes, and hence, they should be ruled out. Commonly known mutations are RET, SDHD, VHL, and SDHB [3]. It is important to identify germline mutations even in patients with no previous family history. It is further worthwhile to screen other family members clinically once testing comes positive [3].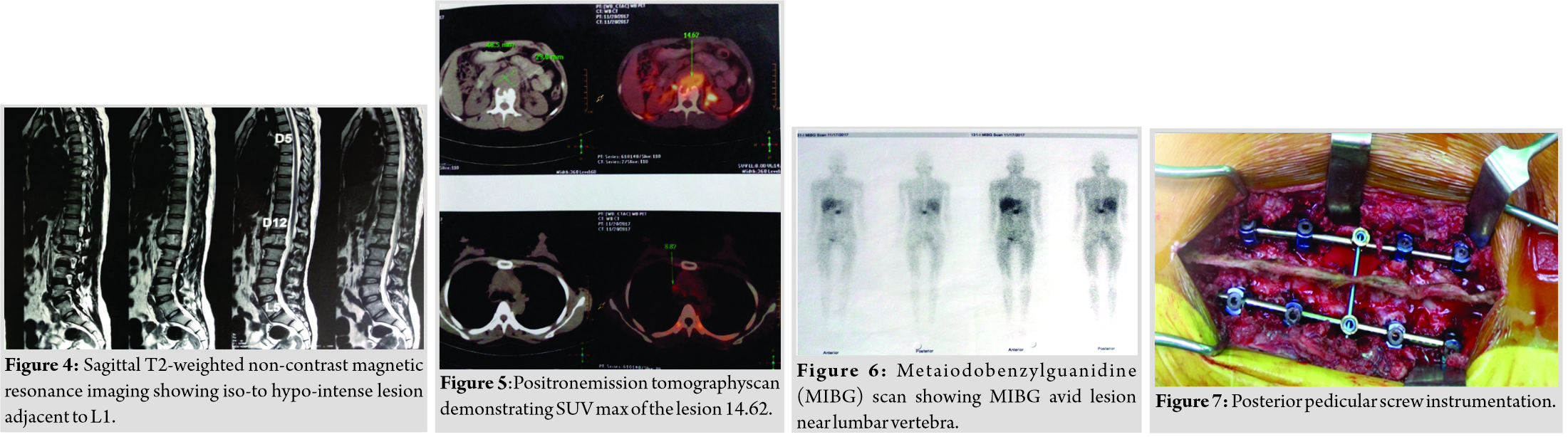
The diagnosis usually starts from blood workup. Phentolamine test, glucagon challenge tests are of historical importance and rarely performed. Since paragangliomas synthesize catecholamines, its metabolites can be found in increased numbers in the plasma and urine. Based on clinical suspicion, the blood and the urine can be tested for one of the elevated catecholamine metabolites [4]. Borderline values usually lead to false-positive diagnosis likely from stress and venipuncture. The sensitivity of plasma metanephrine levels has been reported to be higher than the urinary counterpart and is quoted to be the most sensitive blood marker for diagnosis [4]. Contrast-enhanced CT and MRI are common imaging modalities to be used for diagnosis and localization. On MRI, they show low-to-intermediate signal intensity on T1-weighted images and intermediate to high signal intensity on T2-weighted images [5]. Contrast-enhanced MR images showed intense heterogeneous enhancement which is considered superior to CT in diagnosis. More selective localization of the tumor can be done using radioactive tracers as they exhibit selective uptake in paraganglioma; besides, physiological tissues including 131I- or 123I-MIBG, 111In-somatostatin analogs, or 18F-dopa (or dopamine) PET[5]. Histologically typical cells in nests “Zellballen” pattern arevisible. On immunohistochemistry, tumor cells are positive forchromogranin A suggestive of neuroendocrine differentiation. They are also positive for S-100 and Cyclin-D1 and weakly positive for synaptophysin. One may also find positive markers neuron-specific enolase, cytokeratin, and Vimentin [6]. Cellular atypia, presence of mitoses, and do not necessarily indicate the presence of malignancy. Thus, the term malignant is generally restricted to tumors with distant metastases, most commonly found in lungs, bone, or liver, suggesting a vascular pathway of spread.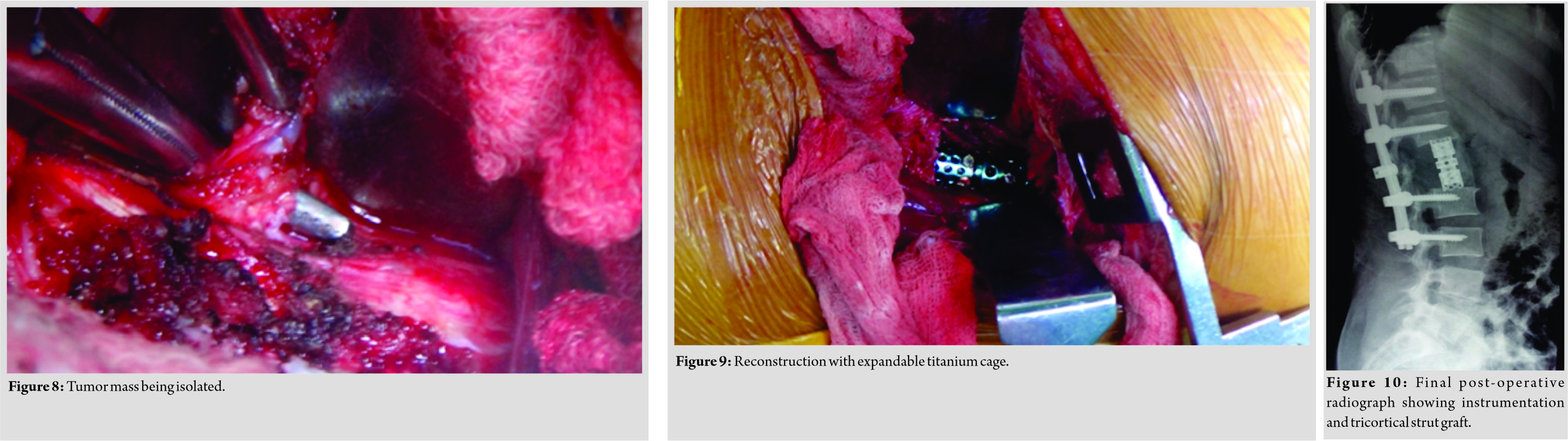
Complete excision of the tumor is necessary to prevent recurrence and is the mainstay of the surgical treatment [6]. Preoperatively, the patient requires introduction of alpha-blocker most commonly phenoxybenzamine for to prevent chronic vagal stone suppression. Usually it is given for a period of 10–14 days. Beta blockers are added once adequate alpha blockade has set in. Intraoperative labile hypertension can be controlled by nitroprusside and hypotension can be managed by restoring volume and inotropes are added if necessary. Our patient required intraoperative inotropes for hypotension. In case of malignancy, 131I-MIBG treatment using 200-mCi doses at monthly intervals, over 3–6 cycles can be given [7].
Conclusion
Lumbar spine secretory paraganglioma needs thorough pre-operative planning and also requires meticulous operative technique and good post-operative monitoring.
Clinical Message
Secretory paragangliomas are uncommon entities and pose difficulty in diagnosis and management. This case report aims to outline the various modalities available to identify the lesion, possible surgical management, and the need for a concurrent team approach for an effective outcome.
References
1. Ozturk B, Coskun U, Yaman E,Akdemir O, Cakir A, Memis L, et al. A case of extra-adrenal paraganglioma localized in retroperitoneum diagnosed with FDG-PET: Case report and review of the literature. Endocrinologist2010;20:60-2.
2. Richter A, Halm HF, Lerner T, Liljenqvist UR, Quante M. Long-term follow-up after en bloc resection and reconstruction of a solitary paraganglioma metastasis in the first lumbar vertebral body: A case report. J Med Case Rep 2011;5:45.
3. Pacak K, Linehan WM, Eisenhofer G, Walther MM, Goldstein DS. Recent advances in genetics, diagnosis, localization, and treatment of pheochromocytoma. Ann Intern Med 2001;134:315-29.
4. Pacak K, Eisenhofer G. An assessment of biochemical tests for the diagnosis of pheochromocytoma. Nat Clin Pract Endocrinol Metab2007;3:744-5.
5. Blake MA, Kalra MK, Maher MM, Sahani DV, Sweeney AT, Mueller PR, et al. Pheochromocytoma: An imaging chameleon. Radiographics 2004;24 Suppl 1:S87-99.
6. Simpson LN, Hughes BD, Karikari IO, Mehta AI, Hodges TR, Cummings TJ, et al. Catecholamine-secreting paraganglioma of the thoracic spinal column: Report of an unusual case and review of the literature. Neurosurgery 2012;70:E1049-52.
7. Sclafani LM, Woodruff JM, Brennan MF. Extraadrenal retroperitoneal paragangliomas: Natural history and response to treatment. Surgery 1990;108:1124-9.
 |
 |
 |
 |
| Dr. Sudhir Srivastava | Dr. Aditya Raj | Dr. Sunil Bhosale | Dr. Nandan Marathe |
| How to Cite This Article: Srivastava S, Raj A, Bhosale S, Marathe N. Secretory Paravertebral Paraganglioma of the Lumbar Spine: A Case Report and Review of Literature. Journal of Orthopaedic Case Reports 2019 Sep-Oct; 9(5): 39-42. |
[Full Text HTML] [Full Text PDF] [XML]
[rate_this_page]
Dear Reader, We are very excited about New Features in JOCR. Please do let us know what you think by Clicking on the Sliding “Feedback Form” button on the <<< left of the page or sending a mail to us at editor.jocr@gmail.com