[box type=”bio”] Learning Point of the Article: [/box]
[box type=”bio”] Learning Point of the Article: [/box]
This is the fifth known case of a locally aggressive, nonrecurrent primary long bone chondroblastoma requiring ambputation, demonstrating the need for early detection, proper diagnosis and follow-up in treating these rare tumors.
Case Report | Volume 10 | Issue 7 | JOCR October 2020 | Page 63-67 | Jonathan Acosta, Jon E. Hammarstedt, Lisa B. Ercolano, Alan Slipak, Gage Trott, Mark J. Sangimin. DOI: 10.13107/jocr.2020.v10.i07.1920
Authors: Jonathan Acosta[1], Jon E. Hammarstedt[1], Lisa B. Ercolano[1], Alan Slipak[1], Gage Trott[1], Mark J. Sangimino[1]
[1]Department of Orthopaedic Surgery, Allegheny General Hospital, Pittsburgh, Pennsylvania, United States.
Address of Correspondence:
Dr. Jon E. Hammarstedt, Allegheny General Hospital,
Department of Orthopaedic Surgery, 1307 Federal Street, 2nd Floor, Pittsburgh PA 15212, United States.
E-mail: jon.hammarstedt@gmail.com
Abstract
Introduction: Chondroblastoma is a rare, cartilaginous primary bone tumor, which presents predominantly in children and young adults. These tumors represent 1% of all primary bone tumors. Patients tend to complain of progressive joint pain which usually aids in early diagnosis. However, early diagnosis has made the understanding of the untreated, progressive course of chondroblastoma quite difficult. This case report highlights a patient who was first encountered with locally advanced chondroblastoma and discusses the challenges of diagnosis and treatment with a focus on the natural progression of this disease.
Case Report: We report the case of a 16-year-old male encountered during relief efforts after the 2010 Haiti earthquake, who was found to have a massive, expansile, and destructive mass of the proximal left tibia and fibula. Radiographic appearance was concerning for a malignant bone forming process. However, biopsy revealed features most consistent with chondroblastoma with secondary aneurysmal bone cyst formation. Marginal resection was considered, but the degree of soft tissue and neurovascular invasion made it impossible to salvage the leg and thus an above-the-knee amputation was performed.
Conclusion: This report reviews the challenging diagnosis of a massive chondroblastoma with locally aggressive features which required ablative surgery. This may provide insight into the untreated, natural course of this pathology.
Keywords: Chondroblastoma, amputation, bone tumor, aggressive.
Introduction
Chondroblastoma is a rare, cartilaginous primary bone tumor, which presents predominantly in children and young adults [1]. These tumors represent 1% of all primary bone tumors. They typically arise in the epiphysis or apophyses of long bones such as the distal femur and proximal tibia and are relatively slow growing. Patients tend to complain of progressive joint pain which usually aids in early diagnosis [1]. Unfortunately, limited health-care resources can prevent patients from accessing treatment in a timely manner. For example, access to care in Haiti in 2010 after the devastating earthquake was virtually nonexistent before foreign aid arrived. We describe the case of a survivor of the Haitian earthquake and the challenging diagnosis and treatment of his chondroblastoma.
Case Report
In 2010, a team of orthopedic surgeons traveled to Haiti to assist in the relief efforts. They encountered a 16-year-old male who had been complaining of painful swelling in his left leg for 10 months. Physical examination revealed a large mass surrounding the left knee with significant soft-tissue swelling. The patient had a 45-degree flexion contracture at the knee and substantial pain with extension. After initial evaluation by the triage team, the patient was transferred to Pittsburgh, PA, USA, for further care.
Initial radiographs revealed a large, expansile mass involving the left proximal tibia and fibula with bony destruction and possible intra-articular extension (Fig. 1). 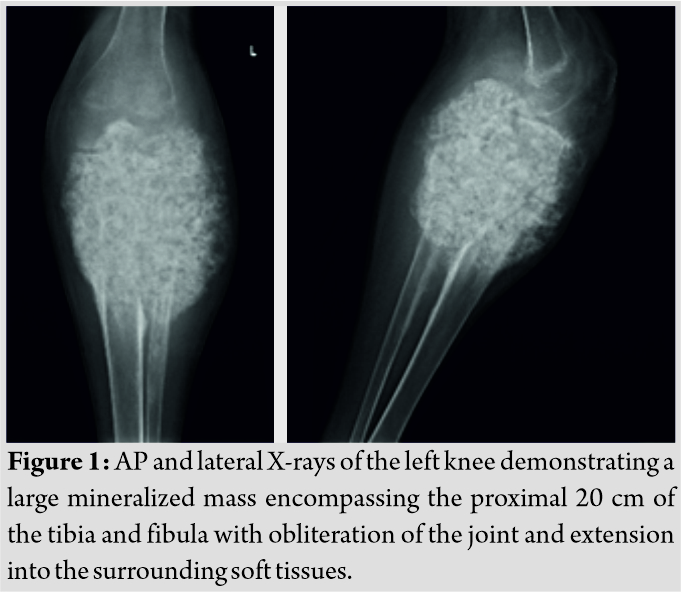 It also demonstrated significant soft-tissue extension, mineralization, and periosteal reaction concerning for osteosarcoma. Computed tomography of the chest, abdomen, pelvis, and spine revealed no evidence of metastatic disease. Magnetic resonance imaging revealed a multilobulated, heterogeneous mass measuring 14 cm × 13 cm × 13 cm replacing the left proximal tibia and fibula, with multicompartmental and intra-articular involvement (Fig. 2, 3).
It also demonstrated significant soft-tissue extension, mineralization, and periosteal reaction concerning for osteosarcoma. Computed tomography of the chest, abdomen, pelvis, and spine revealed no evidence of metastatic disease. Magnetic resonance imaging revealed a multilobulated, heterogeneous mass measuring 14 cm × 13 cm × 13 cm replacing the left proximal tibia and fibula, with multicompartmental and intra-articular involvement (Fig. 2, 3). 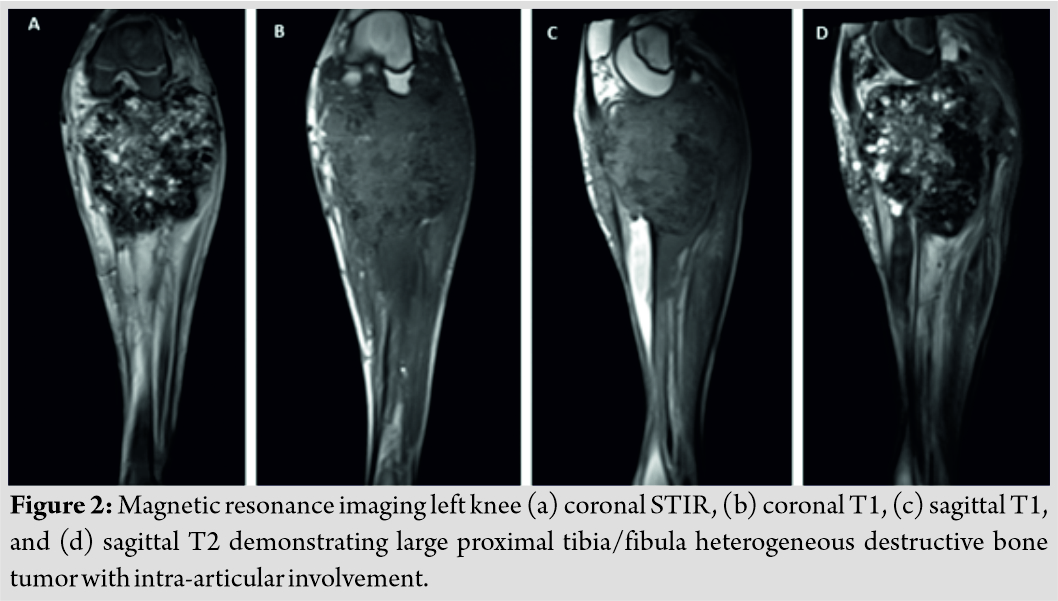 It also showed that the distal popliteal neurovascular bundle was posteriorly displaced with full encasement of the anterior tibial artery at the level of the popliteal trifurcation. In addition, the common peroneal nerve was significantly displaced by the tumor and could not reliably be differentiated from the mass. A magnetic resonance angiogram was performed which showed a hypervascular proximal tibial lesion with poor definition of vascular anatomy. A nuclear medicine bone scan was then performed which showed a large expansile mass of the proximal left tibia, fibula, and distal femur with intense radiotracer uptake. The initial radiographic findings were consistent and suggestive of osteosarcoma, and the patient was subsequently scheduled for biopsy.
It also showed that the distal popliteal neurovascular bundle was posteriorly displaced with full encasement of the anterior tibial artery at the level of the popliteal trifurcation. In addition, the common peroneal nerve was significantly displaced by the tumor and could not reliably be differentiated from the mass. A magnetic resonance angiogram was performed which showed a hypervascular proximal tibial lesion with poor definition of vascular anatomy. A nuclear medicine bone scan was then performed which showed a large expansile mass of the proximal left tibia, fibula, and distal femur with intense radiotracer uptake. The initial radiographic findings were consistent and suggestive of osteosarcoma, and the patient was subsequently scheduled for biopsy.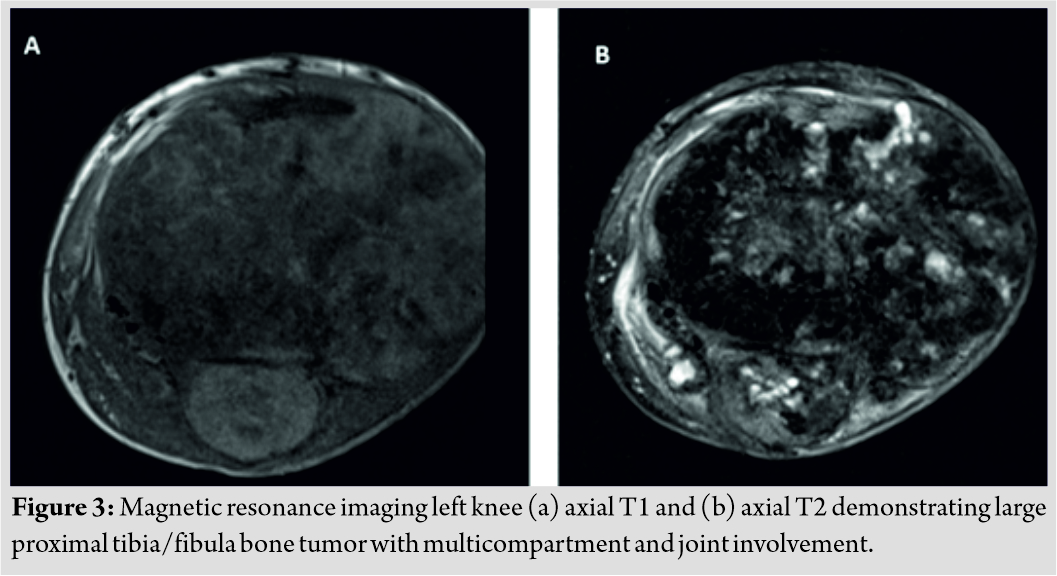
Open incisional biopsy was performed, and the specimen was evaluated by the home institution as well as expert consultation from three outside institutions. Final pathologic assessment determined the mass to be a chondroblastoma with secondary aneurysmal bone cyst formation.
Summary of pathologic assessment
The tumor involved both the tibia and fibula (Fig. 4a). Histologically, the tumor was hypercellular and comprised a population of polygonal cells with eosinophilic cytoplasm and longitudinal nuclear grooves (chondroblasts). Scattered multinucleated osteoclast type giant cells were also present (Fig. 4b). The cells were embedded in a chondroid matrix. Focally, mineralization of the matrix around individual cells imparted a “chicken wire” appearance (Fig. 4c). Focal necrosis and vascular invasion were seen, but marked nuclear atypia and increased mitotic activity were absent (Fig. 4d).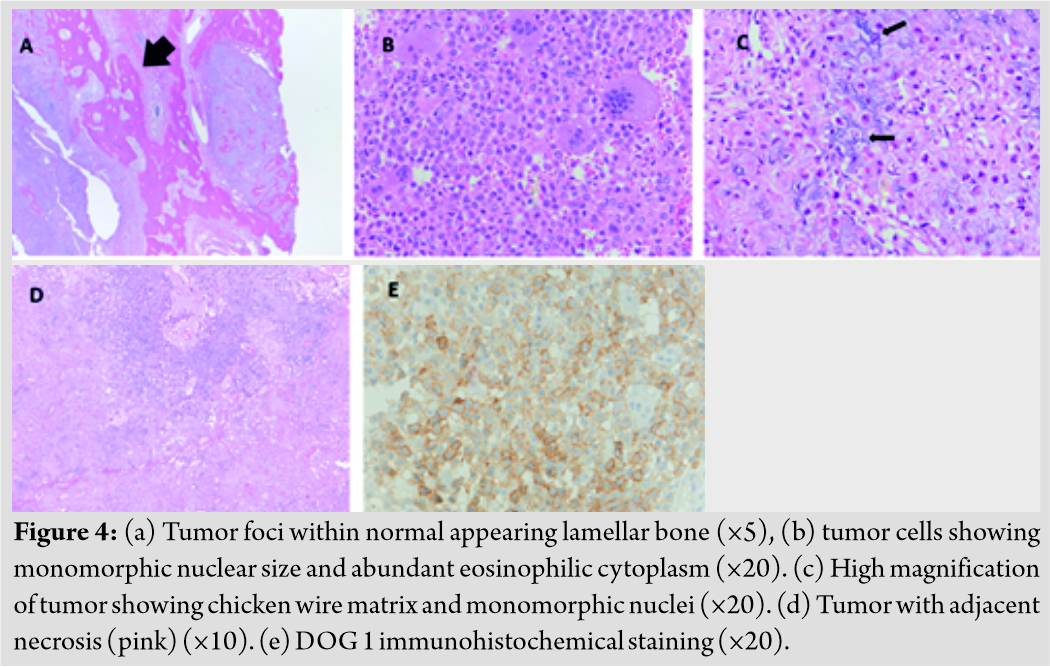
Immunohistochemistry plays a role in the diagnosis of chondroblastoma and the neoplastic cells were positive for S100 protein (a non-specific marker of chondroblasts). With the recent discovery of several sensitive and specific chondroblastoma immunomarkers, the remaining tissue samples were tested for the H3F3 K36M mutation as well as DOG1 immunohistochemistry. The sample tested negative for the H3F3 K36M mutation, but weakly positive for DOG1 (Fig. 4e). Because of the presence of vascular invasion and focal lamellar bone entrapment, expert pathologists considered this lesion to be an atypical chondroblastoma with a secondary aneurysmal bone cyst component. The differential diagnosis at this point also included chondroblastoma-like osteosarcoma but was not favored.
Surgical resection
Marginal resection with knee reconstruction was discussed, but the possibility of above-knee amputation (AKA) was communicated to the patient and family. Intraoperatively, the posterior aspect of the tumor had significantly encased the neurovascular bundle within the popliteal fossa. An intraoperative vascular surgery consult was obtained, and it was determined that chronic pressure from the tumor had caused substantial scarring of the posterior vessels of the leg compromising any chance of restoring flow to the lower leg. The lower extremity had remained viable secondary to formation of collateral vasculature. It was decided that the limb was unsalvageable and not amenable to reconstruction with prosthesis, and thus, an AKA was performed. The excised specimen is shown in Fig. 5.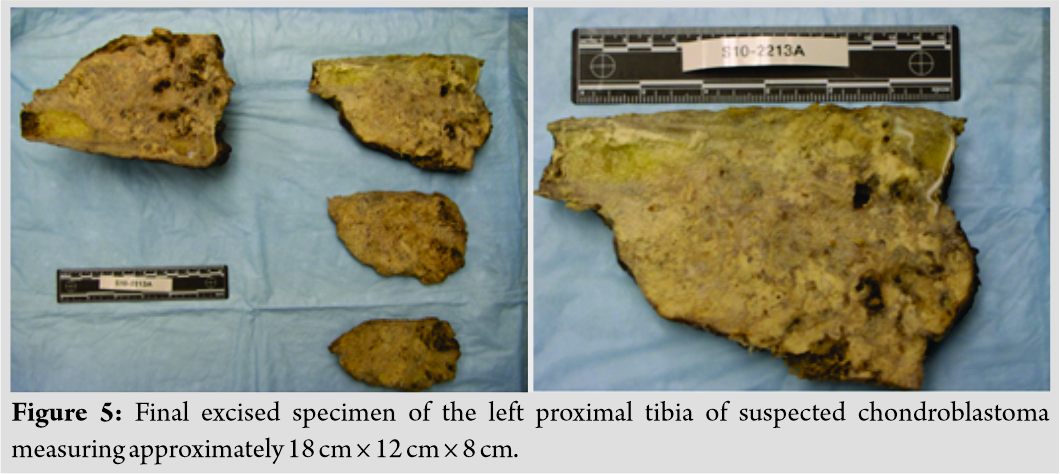
After surgery, the patient was returned to the ICU where the rest of his hospital course was largely unremarkable. He was fitted for a lower leg prosthesis and began physical therapy. After a month spent in the rehabilitation facility, the patient was able to ambulate with crutches and perform most of his activities of daily living unassisted. At latest follow-up of 8 years, the patient has no signs of recurrence or metastasis. He is enrolled in a university, able to perform all activities of daily living, and participates in activities such as soccer with a prosthesis.
Discussion
Several factors contributed to complexity in diagnosis in this case. The first arose during initial imaging. The significant size, soft-tissue mineralization, periosteal reaction, and vascular encasement suggested a malignant etiology such as osteosarcoma. The histologic analysis, however, demonstrated a well-circumscribed and non-infiltrative mass, even with its extensive gross size and significant soft-tissue extension. Morphologically, features were consistent with chondroblastoma and secondary ABC; however, focal necrosis and vascular invasion were noteworthy. With regard to immunohistochemical analysis, the neoplastic cells were S100 positive. Since the treatment of this patient, several new markers have been described which can be useful in difficult cases of suspected chondroblastoma. The first marker is DOG1, which was initially discovered in gastrointestinal stromal tumors but was recently shown to be a marker of chondroblastoma [2]. The second marker, described to be present in 95% of chondroblastomas, is H3F3 K36M mutation which has shown to have a high sensitivity and specificity for diagnosis [3]. However, in this study, 4 of the 77 chondroblastoma cases did not have the K36M mutation, and it is unclear if they identified a different histone variant in those cases. In our case, DOG1 staining was weakly positive, but there was no evidence of the H3F3 K36M mutation. Several factors may have played a role in the results of this immunohistochemical analysis. First, these tests were run approximately 9 years after patient presentation, so the age of the tissues may have caused inaccurate results. Second, the decalcification process of bone tissue can also have a negative influence on immunohistochemistry. Clinically, this lesion was treated as a benign albeit locally aggressive chondroblastoma and with regular follow-up, there has been no evidence of recurrence or distant spread. Although chondroblastomas are benign, wide variability in recurrence rates exist with reported cases of benign metastasis which may lead to a more complicated clinical course. Tumors are typically described as “locally aggressive” when the lesions have significant extracortical invasion and soft-tissue destruction, often a result of late identification and limited intervention. Locally aggressive chondroblastomas are usually associated with local recurrence, which has an incidence of 10%–35% and typically is due to inadequate resection of the primary lesion [1]. Locally aggressive chondroblastomas are associated with higher rates of recurrence [4, 5, 6, 7, 8]. It has been hypothesized that since the majority of locally aggressive cases are associated with recurrence, iatrogenic factors such as curettage-induced cortex breach and leakage may play a significant role in tumor cells accessing the soft tissues [6, 8, 9]. There have been rare cases (>1%) of chondroblastomas which have exhibited metastatic potential and these too have been associated with cases of local recurrence [4, 5, 6, 7, 10]. Unfortunately, the previous attempts have failed at histologically identifying certain features which may predispose chondroblastomas to having an aggressive course. While there is not currently any histological or immunohistochemistry predictor of which lesions will recur or metastasize, these massive and destructive lesions may rightfully be considered higher risk. There have been four previously reported cases of locally aggressive, non-recurrent primary long bone chondroblastoma [11, 12, 13, 14] such as seen in the case of our patient (Table 1). Wright and Sherman [12] described a case of a chondroblastoma of the acromion while Erler et al. [13] reported a case involving the distal femur. In both cases, the tumors demonstrated intra-articular extension with moderate joint effusions present on examination. In another case described by Nouh et al. [13], the patient had a proximal humerus chondroblastoma also initially presenting with a moderate glenohumeral joint effusion and surrounding soft-tissue edema. The case reported by Harish et al. [11] initially presented the most aggressively and most similarly to our patient. Their patient also was diagnosed with a chondroblastoma of the proximal humerus, but its significantly larger size (21 cm ×16 cm ×10 cm), soft-tissue extension, and encasement of axillary vasculature and brachial plexus necessitated a forequarter amputation. Of the four reported non-recurrent locally aggressive chondroblastomas, it was the only reported case before ours requiring amputation due to the significant local soft-tissue extension and neurovascular involvement. Their patient had gone untreated for 12 years, which may have played a significant role in the tumor’s advanced, aggressive nature on presentation. Given the limited access to resources in our patient’s case, it is unclear how long our patient was symptomatic before presentation. It opens the discussion of the natural course of chondroblastomas. Given that only two reported cases exist of this manner, it is critical to answer the question of what factors play a role in determining the behavior of these tumors over their course. Genetic studies of chondroblastomas have revealed a possible association of certain chromosomal abnormalities with metastatic potential [15], but the understanding of what determines the course of this particular tumor remains limited at this time.
Conclusion
Chondroblastomas are benign bone tumors but can be locally aggressive, recur, and rarely metastasize. Our group reports the fifth known case of a locally aggressive, non-recurrent primary long bone chondroblastoma and only the second known instance requiring amputation due to the massive local involvement of tumor. Inherent to its atypical and advanced presentation is the resultant diagnostic difficulty. It is paramount in these circumstances to assure proper diagnosis such that an appropriate treatment course is undertaken. Our specimen was sent to three independent pathologists who were in unanimous agreement of a diagnosis of atypical chondroblastoma with ABC component, despite radiographic considerations, weak staining with DOG1 and negative H3F3 K36M. Amputation was necessary in this case due to local bony destruction and advancement of the tumor relative to vital soft-tissue structures. This case demonstrates the need for early detection, proper diagnosis, and follow-up in treating these rare tumors. Future studies will hopefully elucidate predictive factors of behavior and natural history in these rare tumors.
Clinical Message
This case report describes the second known case of a chondroblastoma requiring amputation and provides further insight into the natural progression of this disease. It also highlights the most recent advancements in genetic and immunohistochemistry diagnostics for improved detection of this rare bone tumor.
References
1. Ramappa AJ, Lee FY, Tang P, Carlson JR, Gebhardt MC, Mankin HJ. Chondroblastoma of bone. J Bone Joint Surg Am 2000;82:1140-5.
2. Akpalo H, Lange C, Zustin J. Discovered on gastrointestinal stromal tumour 1 (DOG1): A useful immunohistochemical marker for diagnosing chondroblastoma. Histopathology 2012;60:1099-106.
3. Amary MF, Berisha F, Mozela R, Gibbons R, Guttridge A, O’Donnell P, et al. The H3F3 K36M mutant antibody is a sensitive and specific marker for the diagnosis of chondroblastoma. Histopathology 2016;69:121-7.
4. Accadbled F, Brouchet A, Salmeron F, Darodes P, Cahuzac JP, Sales De Gauzy J. Recurrent aggressive chondroblastoma: Two cases and a review of the literature. Rev Chir Orthop Reparatr Appar Mot 2001;87:718-23.
5. Kahn LB, Wood FM, Ackerman LV. Malignant chondroblastoma. Report of two cases and reviw of the literature. Arch Pathol 1969;88:371-6.
6. Kyriakos M, Land VJ, Penning HL, Parker SG. Metastatic chondroblastoma. Report of a fatal case with a review of the literature on atypical, aggressive, and malignant chondroblastoma. Cancer 1985;55:1770-89.
7. Mirra JM, Ulich TR, Eckardt JJ, Bhuta S. Aggressive chondroblastoma. Light and ultramicroscopic findings after en bloc resection. Clin Orthop Relat Res 1983;178:276-84.
8. Shoji H, Miller TR. Benign chondroblastoma with soft-tissue recurrence. N Y State J Med 1971;71:2786-9.
9. Coleman SS. Benign chondroblastoma with recurrent soft-tissue and intra-articular lesions. J Bone Joint Surg Am 1966;48:1554-60.
10. Jambhekar NA, Desai PB, Chitale DA, Patil P, Arya S. Benign metastasizing chondroblastoma: A case report. Cancer 1998;82:675-8.
11. Harish K, Janaki MG, Alva NK. Primary aggressive chondroblastoma of the humerus: A case report. BMC Musculoskeletal Disord 2004;5:9.
12. Wright JL, Sherman MS. An unusual chondroblastoma. J Bone Joint Surg Am 1964;46:567-600.
13. Erler K, Yildiz C, Demiralp B, Ozdemir MT, Basbozkurt M. Chondroblastoma of the distal femur. A case report. Acta Orthop Belg 2003;69:467-72.
14. Nouh MR, Abu Shady HM, Abodief WT, Al-Kandary SR. Primary aggressive chondroblastoma of the humerus: An unusual imaging presentation. Clin Imaging 2013;37:783-7.
15. Swarts SJ, Neff JR, Johansson SL, Nelson M, Bridge JA. Significance of abnormalities of chromosomes 5 and 8 in chondroblastoma. Clin Orthop Relat Res 1998;349:189-93.
 |
 |
 |
 |
 |
| Dr. Jonathan Acosta | Dr. Jon E. Hammarstedt | Dr. Lisa B. Ercolano | Dr. Alan Slipak | Dr. Mark J. Sangimino |
| How to Cite This Article: Acosta J, Hammarstedt JE, Ercolano LB, Slipak A, Trott G, Sangimino MJ. A Destructive Tumor of the Proximal Tibia and Fibula: A Case Report Highlighting Challenging Diagnosis and Treatment of a Locally Advanced, Non-recurrent Chondroblastoma in 16 Years Old Patient. Journal of Orthopaedic Case Reports 2020 October;10(7):63-67 |
[Full Text HTML] [Full Text PDF] [XML]
[rate_this_page]
Dear Reader, We are very excited about New Features in JOCR. Please do let us know what you think by Clicking on the Sliding “Feedback Form” button on the <<< left of the page or sending a mail to us at editor.jocr@gmail.com
Related Articles in Journal of Orthopaedic Case Reports
 October 10, 2020 A Destructive Tumor of the Proximal Tibia and Fibula: A Case Report Highlighting Challenging Diagnosis and Treatment of a Locally Advanced, Nonrecurrent Chondroblastoma in a 16 year-old
October 10, 2020 A Destructive Tumor of the Proximal Tibia and Fibula: A Case Report Highlighting Challenging Diagnosis and Treatment of a Locally Advanced, Nonrecurrent Chondroblastoma in a 16 year-old- January 10, 2021 “Dry Arthroscopy” is a Valuable Tool in the Excisional Curettage of Chondroblastoma: A Case Series
 July 1, 2025 Twenty-Four-Year Follow-up of Successful Total Hip Arthroplasty for Recurrent Proximal Femoral Aneurysmal Bone Cyst with a Pathologic Fracture Following Extended Curettage and Bone Grafting: A Case Report
July 1, 2025 Twenty-Four-Year Follow-up of Successful Total Hip Arthroplasty for Recurrent Proximal Femoral Aneurysmal Bone Cyst with a Pathologic Fracture Following Extended Curettage and Bone Grafting: A Case Report July 1, 2025 Chronic Thigh Pain in an Adolescent: A Case Report and Literature Review of Desmoplastic Fibroma, A Rare Benign Tumor
July 1, 2025 Chronic Thigh Pain in an Adolescent: A Case Report and Literature Review of Desmoplastic Fibroma, A Rare Benign Tumor