[box type=”bio”] Learning Point of the Article: [/box]
Spondyloepiphyseal Dysplasia Tarda is a clinically, radiologically distinct syndrome which can be identified on genetic testing which enables definitive diagnosis, prognostication, counseling, management and prenatal diagnosis for such families.
Case Report | Volume 10 | Issue 2 | JOCR March – April 2020 | Page 1-4 | Parag M Tamhankar, Abhishek Kulkarni, Lakshmi Vasudevan. DOI: 10.13107/jocr.2020.v10.i02.1670
Authors: Parag M Tamhankar[1], Abhishek Kulkarni[2], Lakshmi Vasudevan[1]
[1]Centre for Medical Genetics, Ecstasy Business Park, Mumbai, Maharashtra, India,
[2]Endocrine Wellness Clinic, 118 Morya Estate, Mumbai, Maharashtra, India.
Address of Correspondence:
Dr. Parag M Tamhankar,
Centre for Medical Genetics, Office No.250/251, Ecstasy Business Park, JSD Road, Mumbai – 400080, Maharashtra, India.
E-mail: paragtmd2@gmail.com
Abstract
Introduction: X-linked spondyloepiphyseal dysplasia tarda(SEDT) is a type of shorttrunk skeletal dysplasia, occurring in males due to mutation in TRAPPC2 gene.
Case Report: We describe a large Indian family with multiple males affected with X-linked SEDT. The affected individuals presented with disproportionate short stature, short trunk, and barrel-shaped chest. Elder sibs aged 26 years and 31 years had back and hip pain. Premature osteoarthritis was seen requiring hip replacement surgery in one sib. The known pathogenic nonsense mutation c.209G>A (p.W70X) was identified in TRAPPC2 gene. This is the first mutation proven Indian kindred with X-linked SEDT.
Conclusion: Knowledge of molecular basis is essential to provide definitive diagnosis, accurate counseling, and prenatal diagnosis or early postnatal diagnosis for this rare condition.
Keywords: Genetic, India, spondyloepiphyseal dysplasia tarda, TRAPPC2 gene.
Introduction
X-linked spondyloepiphyseal dysplasia tarda (SEDT) (MIM) is a genetic disorder of the vertebral and epiphyseal growth, leading to short stature and degenerative osteoarthropathy. Based on the pattern of inheritance, there are three types, namely, X-linked recessive (MIM 313400), autosomal recessive(MIM 271600), and autosomal dominant (MIM 184100)[1]. The X-linked spondyloepiphyseal tarda is the most common form and mutations are attributed to TRAPPC2 gene found at locus Xp22 [1, 2]. We describe an Indian family with several males affected due to SEDT. Mutations were identified in TRAPPC2 gene in three male sibs and one clinically unaffected carrier sister.
Clinical Summary
Two male sibs born of non-consanguineous parentage, aged 15 years (patient III-6 in Fig.1) and 25 years (patient III-7 in Fig. 1), respectively, were referred for short stature. They were noticed to be lagging behind their peers in height at around 5 years of age. Another maternal cousin aged 31 years (patient III-10, Fig. 1) was also short and had a history of pain in joints of the knee, hip, and the back. He had left-sided partial hip replacement surgery at 30 years of age. Patient III-6 had the following anthropometry: Height 142.5 cm, weight 40 kg, head circumference 57 cm, lower segment (LS) 85 cm, upper segment (US/LS)ratio of 0.67, span 161 cm, and chest circumference 70 cm. Patient III-7had the following anthropometry: Height 147 cm, weight 55 kg, head circumference 58 cm, LS 86 cm, US/LS ratio 0.70, span 166 cm, and chest circumference 98 cm. Patient III-10 had the following anthropometry height 148 cm, weight 58 kg, head circumference 56 cm, LS 87 cm, US/LS ratio 0.70, span 164 cm, and chest circumference 95 cm. All three had barrel-shaped chest, normal facial features, and lumbar lordosis. Patient III-10 also had thoracolumbar scoliosis. Patients III-6 and III-7 did not have any gait defects, joint deformity, swelling, or restricted mobility. They did not have any spinal tenderness or neurological signs. Patient III-10 had restricted hip flexion and abduction and also had a waddling gait. He had spinal tenderness at lower lumbar vertebrae. All patients had normal intelligence. Rest of systems was normal on examination. Radiological survey (Fig. 2) showed platyspondyly, characteristic hump-shaped central and posterior portions of the vertebral bodies in all affected patients. Epiphyses of the femoral head were dysplastic. Joint space of the hip was reduced. Humeral heads were normal. Radiographs of the skull, elbow, hands, and feet were normal. Patient III-10 also had thoracolumbar scoliosis and hip prosthesis was present on the left side. Magnetic resonance imaging spine performed for him showed diffuse flattening of the vertebral bodies, decreased disc spaces, and multilevel disc bulge. Written informed consent was taken from the individuals for genetic testing. The research was conducted in accordance with the principles of Helsinki. Genomic DNA was extracted from peripheral blood. Polymerase chain reaction was performed covering all exons and exon-intron boundaries of the TRAPPC2 gene as per previously described methods [1]. Bidirectional Sanger sequencing was performed on automated capillary sequencer. Mutations are numbered from 1, beginning with the first nucleotide of the translation start [3]. The mutation nomenclatures were as per nucleotide ID NM_001128835.2 and protein ID NP_001122307.2. Patients III-6, III-7, and III-10 were confirmed to be hemizygous for the mutation c.209G>A (p.W70X) in exon 3 of TRAPPC2 gene and individuals II-6, II-9, and III-11 were confirmed as heterozygous carriers for the mutation.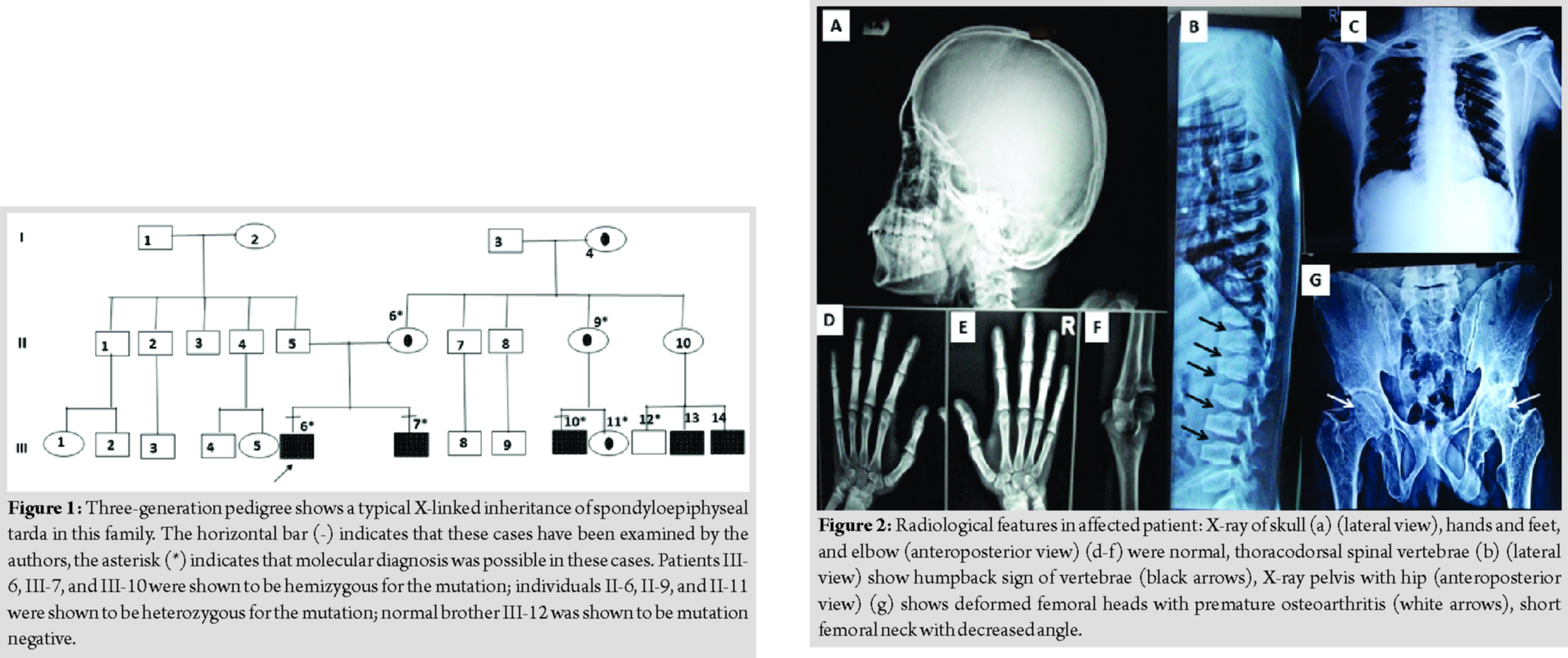
Discussion
Spondyloepiphyseal tardahas been first described by Jacobsen, in 1939 [4]. Gedeon et al. identified the genetic defect in TRAPPC2 gene on locus Xp22 in three unrelated Australian families [5]. The TRAPPC2 gene has six exons and encodes a 140 amino acid protein with putative role in vesicular transport [5]. Several cases of spondyloepiphyseal tarda have been reported from India but none before with molecular confirmation. Pathare et al. reported the first case from Mumbai, Maharashtra, in 1991 [6]. Lakhkar and Raphael reported six unrelated individuals SEDT from Manipal, Karnataka [7]. SEDT does not exhibit any ethnic predisposition. Affected individuals have been described in European, American, Asian, and Australian populations (but not in African-Americans to date) [8]. One estimate suggests that the incidence is two persons per million [9]. The short stature in this family was characterized as disproportionate with short trunk. London Medical Databases lists 74 conditions with short trunk dwarfism. The differential diagnosis is based on clinical presentation and radiological skeletal features. The list of common differential diagnoses includes brachyolmia, spondyloepiphyseal dysplasia congenita, Morquio syndrome, spondyloperipheral dysplasia, multiple epiphyseal dysplasia, and Stickler syndrome [2]. The SEDT form of spondyloepiphyseal dysplasia is milder than the congenita type and is X-linked. Presentation is between 5 and 10 years with back or hip pain like the patients in the present series. Hands, head, and feet appear to be normal size, and final adult height usually ranges from 4’9″ to 5’3″ (range 4’1’’–5’8’’). The SED congenita patients frequently have retinal detachment and cleft palate not seen in SEDT patients. Tiller and Hannig stated that the US/LS ratio is around 0.8 [2]. The US/LS ratio was 0.54 and 0.58 in our patients. Savarirayan et al. stated that there is a great degree of clinical variability even in the same family. The arm span exceeds the height by 10–20 cm and this was observed in our patients too [2, 10]. Characteristically, the vertebrae show a posterior hump of bone on their superior and inferior aspects when viewed laterally [11]. These findings appear in late childhood(after 5 years) but usually before puberty [12]. Anterior beaking of vertebrae can be seen, especially in lower lumbar vertebrae in young patients (Fig. 2). Mild epiphyseal dysplasia is seen in the hips,knees, and shoulders, but rarely in other joints (Fig. 2). Femoral necks are short and bent (coxa vara). Atlantoaxial instability can be presented secondary to odontoid hypoplasia or osodontoideum. Hence, neurological examination is necessary. Scoliosis and lordosis of spine can be progressive warranting treatment such as bracing [2, 8, 12]. The p.W70X mutation observed in our patients has been previously described in other Chinese and British families [13, 14]. However, in the British family, the nucleotide substitution was c.210G>A rather than c.209G>A (seen in our patients and the Chinese family). Fifty-seven unique mutations have been described till date (Human Gene Mutation Database, Cardiff, UK) [15].About 90% of the mutations involve exons 4, 5, and 6 [16].Deletion mutations comprise a third of all mutations. This unusual high deletion frequency, particularly in a gene encoding only a small protein of 140 amino acids, may possibly be explained by the five truncated pseudogenes located on chromosome Yq11.23, which may cause homologous recombination and slipped mispairing [17]. The recurrence risk in pregnancies of carrier females is 50% of males who would be affected. Antenatal ultrasound will not able to diagnose affected fetuses. Due to availability of mutation, genetic counseling, extended family screening, and prenatal diagnosis or early postnatal diagnosis are possible in this family. Treatment remains supportive. Hip replacement is required in several patients with premature degenerative hip arthropathy, during the third decade. Premature arthritis can be prevented by weight management and low impact exercise such as swimming and cycling. In view of possibility atlantoaxial instability, it is prudent to avoid high-risk physical activities such as football, rugby, and trampoline. The patient may require psychosocial counseling for the short stature. Role of growth hormone in increasing final height is not proven [2, 8, 10].
Conclusion
This is the first molecular proven kindred with X-linked SEDT from India. Knowledge of molecular basis is essential to provide definitive diagnosis, accurate counseling, and prenatal diagnosis or early postnatal diagnosis for this rare condition.
Clinical Message
X-linked SEDT is a clinically and radiologically distinguishable genetic syndrome. DNA diagnosis is essential for accurate diagnosis, genetic counseling, prognostication, management, prenatal diagnosis, and early postnatal diagnosis.
References
1. GedeonAK, TillerGE, LeMerrer M, HeuertzS, TranebjaergL, ChitayatD, et al. The molecular basis of X-linked spondyloepiphyseal dysplasia tarda.Am J Hum Genet2001;68:1386-97.
2. Tiller GE, Hannig VL. X-linked spondyloepiphyseal dysplasia tarda. In: Pagon RA, Adam MP, Bird TD, Wallace SE, editors. GeneReviews™. Seattle (WA): University of Washington, Seattle; 2001.
3. denDunnen JT, AntonarakisSE. Mutation nomenclature extensions and suggestions to describe complex mutations: A discussion.Hum Mutat2000;15:7-12.
4. Jacobsen AW. Hereditary osteochondrodystrophia deformans. A family with twenty members affected in five generations. JAMA 1939;113:121-4.
5. GedeonAK, ColleyA, JamiesonR, ThompsonEM, RogersJ, SillenceD, et al. Identification of the gene (SEDL) causing X-linked spondyloepiphyseal dysplasia tarda.Nat Genet1999;22:400-4.
6. Pathare AV, Kothari MA, Chikhalikar AA, Dalvi SG, Vora IM. Spondylo-epiphyseal dysplasia tarda (a case report). J Postgrad Med 1991;37:105-8.
7. Lakhkar BN, Raphael R. Spondyloepiphyseal dysplasia: An evaluation of six cases. Indian J Radiol Imaging 2003;13:199-203.
8. Tiller GE. Spondyloepiphyseal Dysplasia Tarda. National Organisation for Rare diseases. Available from: https://www.rarediseases.org/rare-diseases/spondyloepiphyseal-dysplasia-tarda. [Last accessed on 2019 Oct 01].
9. Wynne-DaviesR, GormleyJ. The prevalence of skeletal dysplasias. An estimate of their minimum frequency and the number of patients requiring orthopaedic care.J Bone Joint Surg Br1985;67:133-7.
10. SavarirayanR, ThompsonE, GéczJ. Spondyloepiphyseal dysplasia tarda (SEDL, MIM #313400).Eur J Hum Genet2003;11:639-42.
11. HarperPS, JenkinsP, LaurenceKM. Spondylo-epiphyseal dysplasia tarda: A report of four cases in two families.Br J Radiol1973;46:676-84.
12. MacKenzieJJ, FitzpatrickJ, BabynP, FerreroGB, BallabioA, BillingsleyG, et al. X linked spondyloepiphyseal dysplasia: A clinical, radiological, and molecular study of a large kindred.J Med Genet1996;33:823-8.
13. ZhuHY, LiJ, ZhuRF, WuX, DuanHL, YangY, et al. Mutational analysis in a family with X-linked spondyloepiphyseal dysplasia tarda.Zhonghua Yi Xue Yi ChuanXue Za Zhi2008;25:421-3.
14. ChristiePT, CurleyA, NesbitMA, ChapmanC, GenetS, HarperPS, et al. Mutational analysis in X-linked spondyloepiphyseal dysplasia tarda.J Clin Endocrinol Metab2001;86:3233-6.
15. StensonPD, MortM, BallEV, ShawK, PhillipsA, CooperDN. The human gene mutation database: Building a comprehensive mutation repository for clinical and molecular genetics, diagnostic testing and personalized genomic medicine.Hum Genet2014;133:1-9.
16. FiedlerJ, LeMerrer M, MortierG, HeuertzS, FaivreL, BrennerRE. X-linked spondyloepiphyseal dysplasia tarda: Novel and recurrent mutations in 13 European families.Hum Mutat2004;24:103.
17. Ryu H, Park J, Chae H, Kim M, Kim Y, Ok IY, et al. X-linked spondyloepiphyseal dysplasia tarda: Identification of a TRAPPC2 mutation in a Korean pedigree. Ann Lab Med 2012;32:234-7.
 |
 |
 |
| Dr. Parag M Tamhankar | Dr. Abhishek Kulkarni | Dr. Lakshmi Vasudevan |
| How to Cite This Article: Tamhankar PM, Kulkarni A, Vasudevan L. X-linked Spondyloepiphyseal Dysplasia Tarda with Mutation in TRAPPC2 Gene: First Report from India. Journal of Orthopaedic Case Reports 2020 Mar-Apr;10(2): 1-4. |
[Full Text HTML] [Full Text PDF] [XML]
[rate_this_page]
Dear Reader, We are very excited about New Features in JOCR. Please do let us know what you think by Clicking on the Sliding “Feedback Form” button on the <<< left of the page or sending a mail to us at editor.jocr@gmail.com